
Atomic-scale studies of catalysis
by spinel oxides
Subproject P04
The spinel class of metal oxides hosts diverse materials, some of which make excellent catalysts. Fe3O4 is already the industrial catalyst for the high-temperature water-gas shift reaction (CO+H2O -> H2+CO2), but research is needed to identify the optimal replacement for the toxic Cr promoter/stabilizer. Ternary MeFe2O4 compounds (Me=Fe, Ni, Co, Mn) are active and stable for the electrochemical oxygen evolution reaction (OER). However, the structure of the active catalyst and the reaction mechanisms are unknown. While these reactions appear different, both clearly benefit from a combination of multivalent cations in the surface layers.
In this project, we will seek to learn why, using a combination of atomic-scale imaging, a host of spectroscopies, and theory. We will dope the Fe3O4(001) surface with 3d transition metals and investigate how the adsorption energies, XPS binding energies, and IRAS frequencies of H2O, CO, CO2, O2, and H2 change with sample composition all the way from isolated dopants to ternary thin films. We will use the data obtained in tightly-controlled UHV experiments to:
i) Interpret the reactivity of our model catalysts under realistic HTWGS and OER conditions.
ii) Provide the benchmark data for experiments on nominally similar powder catalysts (P10 Föttinger).
iii) Support the development of theoretical modeling (P07 Franchini).
A joint postdoc (P04-P11) will facilitate the new collaboration with P11 Backus.
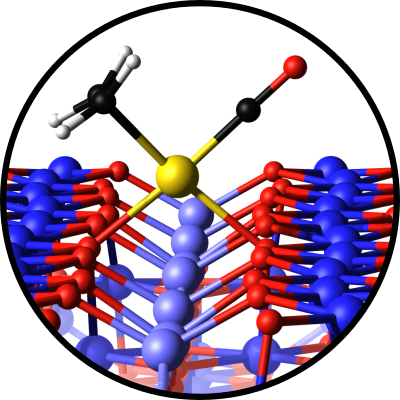

Gareth S. Parkinson
PI
Expertise
- Scanning Tunneling Microscopy (STM) (in UHV 4K – 300 K, electrochemical STM)
- Atomic Force Microscopy (AFM): UHV-based (q+ sensor) and in the ambient (cantilever-based)
- Low-Energy Electron Diffraction (LEED)
- Reflection High Energy Diffraction (RHEED)
- X-ray Photoelectron Spectroscopy (XPS)
- Ultraviolet Photoelectron Spectroscopy (UPS)
- Auger Electron Spectroscopy (AES)
- Low-energy He+ ion scattering (LEIS)
- Thermal Programmed Desorption Spectroscopy (TPD)
Team

Gareth S. Parkinson
PI

Jiří Pavelec
co-PI

Chunlei Wang
PostDoc

David Rath
PhD Student
Associates

Moritz Eder
PostDoc
Publications
2024
Wang, Chunlei; Sombut, Panukorn; Puntscher, Lena; Jakub, Zdenek; Meier, Matthias; Pavelec, Jiri; Bliem, Roland; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare; Parkinson, Gareth S.
CO‐Induced Dimer Decay Responsible for Gem‐Dicarbonyl Formation on a Model Single‐Atom Catalyst
Journal ArticleOpen AccessIn PressIn: Angewandte Chemie - International Edition, no. e202317347, 2024, ISSN: 1521-3773.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Wang2024,
title = {CO‐Induced Dimer Decay Responsible for Gem‐Dicarbonyl Formation on a Model Single‐Atom Catalyst},
author = {Chunlei Wang and Panukorn Sombut and Lena Puntscher and Zdenek Jakub and Matthias Meier and Jiri Pavelec and Roland Bliem and Michael Schmid and Ulrike Diebold and Cesare Franchini and Gareth S. Parkinson},
doi = {10.1002/anie.202317347},
issn = {1521-3773},
year = {2024},
date = {2024-01-31},
journal = {Angewandte Chemie - International Edition},
number = {e202317347},
publisher = {Wiley},
abstract = {The ability to coordinate multiple reactants at the same active site is important for the wide-spread applicability of single-atom catalysis. Model catalysts are ideal to investigate the link between active site geometry and reactant binding, because the structure of single-crystal surfaces can be precisely determined, the adsorbates imaged by scanning tunneling microscopy (STM), and direct comparisons made to density functional theory. In this study, we follow the evolution of Rh_{1} adatoms and minority Rh_{2} dimers on Fe_{3}O_{4}(001) during exposure to CO using time-lapse STM at room temperature. CO adsorption at Rh_{1} sites results exclusively in stable Rh_{1}CO monocarbonyls, because the Rh atom adapts its coordination to create a stable pseudo-square planar environment. Rh_{1}(CO)_{2} gem-dicarbonyl species are also observed, but these form exclusively through the breakup of Rh_{2} dimers via an unstable Rh_{2}(CO)_{3} intermediate. Overall, our results illustrate how minority species invisible to area-averaging spectra can play an important role in catalytic systems, and show that the decomposition of dimers or small clusters can be an avenue to produce reactive, metastable configurations in single-atom catalysis.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Birschitzky, Viktor; Sokolovic, Igor; Prezzi, Michael; Palotas, Krisztian; Setvin, Martin; Diebold, Ulrike; Reticcioli, Michele; Franchini, Cesare
Machine Learning Based Prediction of Polaron-Vacancy Patterns on the TiO2(110) Surface
Journal ArticleOpen AccessAccepted ArticlearXivIn: npj Computational Materials, 2024.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Birschitzky_2024a,
title = {Machine Learning Based Prediction of Polaron-Vacancy Patterns on the TiO2(110) Surface},
author = {Viktor Birschitzky and Igor Sokolovic and Michael Prezzi and Krisztian Palotas and Martin Setvin and Ulrike Diebold and Michele Reticcioli and Cesare Franchini},
doi = { https://doi.org/10.48550/arXiv.2401.12042},
year = {2024},
date = {2024-01-22},
journal = {npj Computational Materials},
abstract = {The multifaceted physics of oxides is shaped by their composition and the presence of defects, which are often accompanied by the formation of polarons. The simultaneous presence of polarons and defects, and their complex interactions, pose challenges for first-principles simulations and experimental techniques. In this study, we leverage machine learning and a first-principles database to analyze the distribution of surface oxygen vacancies (VO) and induced small polarons on rutile TiO2(110), effectively disentangling the interactions between polarons and defects. By combining neural-network supervised learning and simulated annealing, we elucidate the inhomogeneous VO distribution observed in scanning probe microscopy (SPM). Our innovative approach allows us to understand and predict defective surface patterns at previously inaccessible length scales, identifying the specific role of individual types of defects. Specifically, surface-polaron-stabilizing VO-configurations are identified, which could have consequences for surface reactivity.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
2023
Puntscher, Lena; Sombut, Panukorn; Wang, Chunlei; Ulreich, Manuel; Pavelec, Jiri; Rafsanjani-Abbasi, Ali; Meier, Matthias; Lagin, Adam; Setvin, Martin; Diebold, Ulrike; Franchini, Cesare; Schmid, Michael; Parkinson, Gareth S.
A Multitechnique Study of C2H4 Adsorption on Fe3O4(001)
Journal ArticleOpen AccessIn: Journal of Physical Chemistry C, vol. 127, iss. 37, pp. 18378–18388, 2023.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Puntscher2023,
title = {A Multitechnique Study of C_{2}H_{4} Adsorption on Fe_{3}O_{4}(001)},
author = {Lena Puntscher and Panukorn Sombut and Chunlei Wang and Manuel Ulreich and Jiri Pavelec and Ali Rafsanjani-Abbasi and Matthias Meier and Adam Lagin and Martin Setvin and Ulrike Diebold and Cesare Franchini and Michael Schmid and Gareth S. Parkinson},
doi = {10.1021/acs.jpcc.3c03684},
year = {2023},
date = {2023-09-11},
urldate = {2023-09-11},
journal = {Journal of Physical Chemistry C},
volume = {127},
issue = {37},
pages = {18378--18388},
publisher = {American Chemical Society (ACS)},
abstract = {The adsorption/desorption of ethene (C_{2}H_{4}), also commonly known as ethylene, on Fe_{3}O_{4}(001) was studied under ultrahigh vacuum conditions using temperature-programmed desorption (TPD), scanning tunneling microscopy, X-ray photoelectron spectroscopy, and density functional theory (DFT)-based computations. To interpret the TPD data, we have employed a new analysis method based on equilibrium thermodynamics. C_{2}H_{4} adsorbs intact at all coverages and interacts most strongly with surface defects such as antiphase domain boundaries and Fe adatoms. On the regular surface, C_{2}H_{4} binds atop surface Fe sites up to a coverage of 2 molecules per (√2 × √2)R45° unit cell, with every second Fe occupied. A desorption energy of 0.36 eV is determined by analysis of the TPD spectra at this coverage, which is approximately 0.1–0.2 eV lower than the value calculated by DFT + U with van der Waals corrections. Additional molecules are accommodated in between the Fe rows. These are stabilized by attractive interactions with the molecules adsorbed at Fe sites. The total capacity of the surface for C_{2}H_{4} adsorption is found to be close to 4 molecules per (√2 × √2)R45° unit cell.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Kraushofer, Florian; Meier, Matthias; Jakub, Zdeněk; Hütner, Johanna; Balajka, Jan; Hulva, Jan; Schmid, Michael; Franchini, Cesare; Diebold, Ulrike; Parkinson, Gareth S.
Oxygen-Terminated (1 × 1) Reconstruction of Reduced Magnetite Fe3O4(111)
Journal ArticleOpen AccessIn: vol. 14, no. 13, pp. 3258–3265, 2023.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Kraushofer2023,
title = {Oxygen-Terminated (1 × 1) Reconstruction of Reduced Magnetite Fe_{3}O_{4}(111)},
author = {Florian Kraushofer and Matthias Meier and Zdeněk Jakub and Johanna Hütner and Jan Balajka and Jan Hulva and Michael Schmid and Cesare Franchini and Ulrike Diebold and Gareth S. Parkinson},
doi = {10.1021/acs.jpclett.3c00281},
year = {2023},
date = {2023-03-28},
urldate = {2023-03-28},
volume = {14},
number = {13},
pages = {3258--3265},
publisher = {American Chemical Society (ACS)},
abstract = {The (111) facet of magnetite (Fe_{3}O_{4}) has been studied extensively by experimental and theoretical methods, but controversy remains regarding the structure of its low-energy surface terminations. Using density functional theory (DFT) computations, we demonstrate three reconstructions that are more favorable than the accepted Feoct2 termination under reducing conditions. All three structures change the coordination of iron in the kagome Feoct1 layer to be tetrahedral. With atomically resolved microscopy techniques, we show that the termination that coexists with the Fetet1 termination consists of tetrahedral iron capped by 3-fold coordinated oxygen atoms. This structure explains the inert nature of the reduced patches.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Verdi, Carla; Ranalli, Luigi; Franchini, Cesare; Kresse, Georg
Journal ArticleIn: Physical Review Materials, vol. 7, no. 3, pp. l030801, 2023.
Abstract | Links | BibTeX | Tags: P03, P07
@article{Verdi2023,
title = {Quantum paraelectricity and structural phase transitions in strontium titanate beyond density functional theory},
author = {Carla Verdi and Luigi Ranalli and Cesare Franchini and Georg Kresse},
doi = {10.1103/physrevmaterials.7.l030801},
year = {2023},
date = {2023-03-16},
journal = {Physical Review Materials},
volume = {7},
number = {3},
pages = {l030801},
publisher = {American Physical Society (APS)},
abstract = {We demonstrate an approach for calculating temperature-dependent quantum and anharmonic effects with beyond density-functional theory accuracy. By combining machine-learned potentials and the stochastic self-consistent harmonic approximation, we investigate the cubic to tetragonal transition in strontium titanate and show that the paraelectric phase is stabilized by anharmonic quantum fluctuations. We find that a quantitative understanding of the quantum paraelectric behavior requires a higher-level treatment of electronic correlation effects via the random phase approximation. This approach enables detailed studies of emergent properties in strongly anharmonic materials beyond density-functional theory.},
keywords = {P03, P07},
pubstate = {published},
tppubtype = {article}
}
Ranalli, Luigi; Verdi, Carla; Monacelli, Lorenzo; Kresse, Georg; Calandra, Matteo; Franchini, Cesare
Journal ArticleOpen AccessIn: Advanced Quantum Technology, vol. 6, iss. 4, 2023.
Abstract | Links | BibTeX | Tags: P03, P07
@article{Ranalli2023,
title = {Temperature-dependent anharmonic phonons in quantum paraelectric KTaO_{3} by first principles and machine-learned force fields},
author = {Luigi Ranalli and Carla Verdi and Lorenzo Monacelli and Georg Kresse and Matteo Calandra and Cesare Franchini},
doi = {10.1002/qute.202200131},
year = {2023},
date = {2023-02-22},
urldate = {2023-02-22},
journal = {Advanced Quantum Technology},
volume = {6},
issue = {4},
abstract = {Understanding collective phenomena in quantum materials from first principles is a promising route toward engineering materials properties and designing new functionalities. This work examines the quantum paraelectric state, an elusive state of matter characterized by the smooth saturation of the ferroelectric instability at low temperature due to quantum fluctuations associated with anharmonic phonon effects. The temperature-dependent evolution of the soft ferroelectric phonon mode in the quantum paraelectric KTaO_{3} in the range 0–300 K is modeled by combining density functional theory (DFT) calculations with the stochastic self-consistent harmonic approximation assisted by an on-the-fly machine-learned force field. The calculated data show that including anharmonic terms is essential to stabilize the spurious imaginary ferroelectric phonon predicted by DFT in the harmonic approximation, in agreement with experiments. Augmenting the DFT workflow with machine-learned force fields allows for efficient stochastic sampling of the configuration space using large supercells in a wide temperature range, inaccessible to conventional ab initio protocols. This work proposes a robust computational workflow capable of accounting for collective behaviors involving different degrees of freedom and occurring at large time/length scales, paving the way for precise modeling and control of quantum effects in materials.},
keywords = {P03, P07},
pubstate = {published},
tppubtype = {article}
}
Corrias, Marco; Papa, Lorenzo; Sokolovíc, Igor; Birschitzky, Viktor; Gorfer, Alexander; Setvin, Martin; Schmid, Michael; Diebold, Ulrike; Reticcioli, Michele; Franchini, Cesare
Automated Real-Space Lattice Extraction for Atomic Force Microscopy Images
Journal ArticleOpen AccessIn: Machine Learning: Science and Technology, vol. 4, pp. 015015, 2023.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Corrias2023,
title = {Automated Real-Space Lattice Extraction for Atomic Force Microscopy Images},
author = {Marco Corrias and Lorenzo Papa and Igor Sokolovíc and Viktor Birschitzky and Alexander Gorfer and Martin Setvin and Michael Schmid and Ulrike Diebold and Michele Reticcioli and Cesare Franchini},
doi = {10.1088/2632-2153/acb5e0},
year = {2023},
date = {2023-01-24},
urldate = {2023-01-24},
journal = {Machine Learning: Science and Technology},
volume = {4},
pages = {015015},
abstract = {Analyzing atomically resolved images is a time-consuming process requiring solid experience and substantial human intervention. In addition, the acquired images contain a large amount of information such as crystal structure, presence and distribution of defects, and formation of domains, which need to be resolved to understand a material's surface structure. Therefore, machine learning techniques have been applied in scanning probe and electron microscopies during the last years, aiming for automatized and efficient image analysis. This work introduces a free and open source tool (AiSurf: Automated Identification of Surface Images) developed to inspect atomically resolved images via Scale-Invariant Feature Transform (SIFT) and Clustering Algorithms (CA). AiSurf extracts primitive lattice vectors, unit cells, and structural distortions from the original image, with no pre-assumption on the lattice and minimal user intervention. The method is applied to various atomically resolved non-contact atomic force microscopy (AFM) images of selected surfaces with different levels of complexity: anatase TiO_{2}(101), oxygen deficient rutile TiO_{2}(110) with and without CO adsorbates, SrTiO_{3}(001) with Sr vacancies and graphene with C vacancies. The code delivers excellent results and is tested against atom misclassification and artifacts, thereby facilitating the interpretation of scanning probe microscopy images.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
2022
Wang, Zhichang; Reticcioli, Michele; Jakub, Zdenek; Sokolović, Igor; Meier, Matthias; Boatner, Lynn A; Schmid, Michael; Parkinson, Gareth S.; Diebold, Ulrike; Franchini, Cesare; Setvin, Martin
Surface chemistry on a polarizable surface: Coupling of CO with KTaO 3(001)
Journal ArticleOpen AccessIn: Science Advances, vol. 8, iss. 33, 2022.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Wang2022,
title = {Surface chemistry on a polarizable surface: Coupling of CO with KTaO _{3}(001)},
author = {Zhichang Wang and Michele Reticcioli and Zdenek Jakub and Igor Sokolović and Matthias Meier and Lynn A Boatner and Michael Schmid and Gareth S. Parkinson and Ulrike Diebold and Cesare Franchini and Martin Setvin},
url = {https://www.science.org/doi/10.1126/sciadv.abq1433},
doi = {10.1126/sciadv.abq1433},
year = {2022},
date = {2022-08-19},
urldate = {2022-08-19},
journal = {Science Advances},
volume = {8},
issue = {33},
publisher = {American Association for the Advancement of Science (AAAS)},
abstract = {Polarizable materials attract attention in catalysis because they have a free parameter for tuning chemical reactivity. Their surfaces entangle the dielectric polarization with surface polarity, excess charge, and orbital hybridization. How this affects individual adsorbed molecules is shown for the incipient ferroelectric perovskite KTaO_{3}. This intrinsically polar material cleaves along (001) into KO- and TaO_{2}-terminated surface domains. At TaO_{2} terraces, the polarity-compensating excess electrons form a two-dimensional electron gas and can also localize by coupling to ferroelectric distortions. TaO_{2} terraces host two distinct types of CO molecules, adsorbed at equivalent lattice sites but charged differently as seen in atomic force microscopy/scanning tunneling microscopy. Temperature-programmed desorption shows substantially stronger binding of the charged CO; in density functional theory calculations, the excess charge favors a bipolaronic configuration coupled to the CO. These results pinpoint how adsorption states couple to ferroelectric polarization.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Reticcioli, Michele; Wang, Zhichang; Schmid, Michael; Wrana, Dominik; Boatner, Lynn A.; Diebold, Ulrike; Setvin, Martin; Franchini, Cesare
Competing electronic states emerging on polar surfaces
Journal ArticleOpen AccessIn: Nature Communications, vol. 13, no. 4311, 2022.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Reticcioli2022,
title = {Competing electronic states emerging on polar surfaces},
author = {Michele Reticcioli and Zhichang Wang and Michael Schmid and Dominik Wrana and Lynn A. Boatner and Ulrike Diebold and Martin Setvin and Cesare Franchini},
url = {https://www.nature.com/articles/s41467-022-31953-6},
doi = {10.1038/s41467-022-31953-6},
year = {2022},
date = {2022-07-25},
urldate = {2022-07-25},
journal = {Nature Communications},
volume = {13},
number = {4311},
publisher = {Springer Science and Business Media LLC},
abstract = {Excess charge on polar surfaces of ionic compounds is commonly described by the two-dimensional electron gas (2DEG) model, a homogeneous distribution of charge, spatially-confined in a few atomic layers. Here, by combining scanning probe microscopy with density functional theory calculations, we show that excess charge on the polar TaO_{2} termination of KTaO_{3}(001) forms more complex electronic states with different degrees of spatial and electronic localization: charge density waves (CDW) coexist with strongly-localized electron polarons and bipolarons. These surface electronic reconstructions, originating from the combined action of electron-lattice interaction and electronic correlation, are energetically more favorable than the 2DEG solution. They exhibit distinct spectroscopy signals and impact on the surface properties, as manifested by a local suppression of ferroelectric distortions.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
![Role of Polarons in Single-Atom Catalysts: Case Study of Me1[Au1,Pt1 and Rh1] on TiO2(110)](https://sfb-taco.at/wp-content/uploads/2023/02/P07_P04-300x300.png)
Sombut, Panukorn; Puntscher, Lena; Atzmüller, Marlene; Jakub, Zdenek; Reticcioli, Michele; Meier, Matthias; Parkinson, Gareth S.; Franchini, Cesare
Role of Polarons in Single-Atom Catalysts: Case Study of Me1[Au1,Pt1 and Rh1] on TiO2(110)
Journal ArticleOpen AccessIn: Topics in Catalysis, vol. 65, pp. 1620–1630, 2022.
Abstract | Links | BibTeX | Tags: P04, P07
@article{Sombut2022,
title = {Role of Polarons in Single-Atom Catalysts: Case Study of Me_{1}[Au_{1},Pt_{1} and Rh_{1}] on TiO_{2}(110)},
author = {Panukorn Sombut and Lena Puntscher and Marlene Atzmüller and Zdenek Jakub and Michele Reticcioli and Matthias Meier and Gareth S. Parkinson and Cesare Franchini},
doi = {10.1007/s11244-022-01651-0},
year = {2022},
date = {2022-07-25},
journal = {Topics in Catalysis},
volume = {65},
pages = {1620--1630},
abstract = {The local environment of metal-oxide supported single-atom catalysts plays a decisive role in the surface reactivity and related catalytic properties. The study of such systems is complicated by the presence of point defects on the surface, which are often associated with the localization of excess charge in the form of polarons. This can affect the stability, the electronic configuration, and the local geometry of the adsorbed adatoms. In this work, through the use of density functional theory and surface-sensitive experiments, we study the adsorption of Rh_{1}, Pt_{1}, and Au_{1} metals on the reduced TiO_{2}(110) surface, a prototypical polaronic material. A systematic analysis of the adsorption configurations and oxidation states of the adsorbed metals reveals different types of couplings between adsorbates and polarons. As confirmed by scanning tunneling microscopy measurements, the favored Pt_{1} and Au_{1} adsorption at oxygen vacancy sites is associated with a strong electronic charge transfer from polaronic states to adatom orbitals, which results in a reduction of the adsorbed metal. In contrast, the Rh_{1} adatoms interact weakly with the excess charge, which leaves the polarons largely unaffected. Our results show that an accurate understanding of the properties of single-atom catalysts on oxide surfaces requires a careful account of the interplay between adatoms, vacancy sites, and polarons.},
keywords = {P04, P07},
pubstate = {published},
tppubtype = {article}
}