
Publications
2024
Wanzenböck, Ralf; Buchner, Florian; Kovács, Péter; Madsen, Georg K. H.; Carrete, Jesús
Clinamen2: Functional-style evolutionary optimization in Python for atomistic structure searches
Journal ArticleForthcomingOpen AccessIn: Computer Physics Communications, vol. 297, no. 109065, Forthcoming.
Abstract | Links | BibTeX | Tags: P09
@article{wanzenboeck2024,
title = {Clinamen2: Functional-style evolutionary optimization in Python for atomistic structure searches},
author = {Ralf Wanzenböck and Florian Buchner and Péter Kovács and Georg K. H. Madsen and Jesús Carrete},
doi = {10.1016/j.cpc.2023.109065},
year = {2024},
date = {2024-04-30},
urldate = {2024-04-01},
journal = {Computer Physics Communications},
volume = {297},
number = {109065},
abstract = {Clinamen2 is a versatile functional-style Python implementation of the covariance matrix adaptation evolution strategy (CMA-ES) utilizing Cholesky decomposition. On top of a problem-agnostic core algorithm, the software package offers a suite of utilities and library code enabling applications to important atomistic structure searches. Features include massively distributed computation and the BI-Population restart scheme. This article details the general code structure and introduces examples that illustrate some relevant applications for the materials science and chemistry worlds, including interfacing to density-functional-theory codes and machine-learned surrogate models. The functional design renders the code modular and adaptable, and makes the creation of interfaces to other atomistic software straightforward.},
keywords = {P09},
pubstate = {forthcoming},
tppubtype = {article}
}
Rath, David; Mikerásek, Vojtěch; Wang, Chunlei; Eder, Moritz; Schmid, Michael; Diebold, Ulrike; Parkinson, Gareth S.; Pavelec, Jiří
Journal ArticleOpen AccessSubmittedarXivIn: arXiv, 2024.
Abstract | Links | BibTeX | Tags: P02, P04
@article{Rath2024,
title = {Infrared Reflection Absorption Spectroscopy Setup with Incidence Angle Selection for Surfaces of Non-Metals},
author = {David Rath and Vojtěch Mikerásek and Chunlei Wang and Moritz Eder and Michael Schmid and Ulrike Diebold and Gareth S. Parkinson and Jiří Pavelec},
url = {https://arxiv.org/abs/2403.19263},
doi = {10.48550/ARXIV.2403.19263},
year = {2024},
date = {2024-03-28},
journal = {arXiv},
publisher = {arXiv},
abstract = {Infrared Reflection Absorption Spectroscopy (IRAS) on dielectric single crystals is challenging because the optimal incidence angles for light-adsorbate interaction coincide with regions of low IR reflectivity. Here, we introduce an optimized IRAS setup that maximizes the signal-to-noise ratio for non-metals. This is achieved by maximizing light throughput, and by selecting optimal incidence angles that directly impact the peak heights in the spectra. The setup uses a commercial FTIR spectrometer and is usable in ultra-high vacuum (UHV). Specifically, the design features sample illumination and collection mirrors with a high numerical aperture inside the UHV system, and an adjustable aperture to select the incidence angle range on the sample. This is important for p-polarized measurements on dielectrics, because the peaks in the spectra reverse direction at the Brewster angle (band inversion). The system components are connected precisely via a single flange, ensuring long-term stability. We studied the signal-to-noise (SNR) variation in p-polarized IRAS spectra for one monolayer of CO on TiO_{2}(110) as a function of incidence angle range, where a maximum signal-to-noise ratio of 70 was achieved at 4 cm^{-1} resolution in five minutes measurement time. The capabilities for s-polarization are demonstrated by measuring one monolayer D_{2}O adsorbed on a TiO_{2}(110) surface, where a SNR of 65 was achieved at a delta_R/R0 peak height of 1.4x10-4 in twenty minutes.},
keywords = {P02, P04},
pubstate = {published},
tppubtype = {article}
}
de Hijes, Pablo Montero; Dellago, Christoph; Jinnouchi, Ryosuke; Schmiedmayer, Bernhard; Kresse, Georg
Journal ArticleOpen AccessIn: The Journal of Chemical Physics, vol. 160, iss. 11, no. 114107, 2024.
Abstract | Links | BibTeX | Tags: P03, P12
@article{10.1063/5.0197105,
title = {Comparing machine learning potentials for water: Kernel-based regression and Behler–Parrinello neural networks},
author = {Pablo Montero de Hijes and Christoph Dellago and Ryosuke Jinnouchi and Bernhard Schmiedmayer and Georg Kresse},
doi = {https://doi.org/10.1063/5.0197105},
year = {2024},
date = {2024-03-20},
urldate = {2024-03-20},
journal = {The Journal of Chemical Physics},
volume = {160},
number = {114107},
issue = {11},
abstract = {In this paper, we investigate the performance of different machine learning potentials (MLPs) in predicting key thermodynamic properties of water using RPBE + D3. Specifically, we scrutinize kernel-based regression and high-dimensional neural networks trained on a highly accurate dataset consisting of about 1500 structures, as well as a smaller dataset, about half the size, obtained using only on-the-fly learning. This study reveals that despite minor differences between the MLPs, their agreement on observables such as the diffusion constant and pair-correlation functions is excellent, especially for the large training dataset. Variations in the predicted density isobars, albeit somewhat larger, are also acceptable, particularly given the errors inherent to approximate density functional theory. Overall, this study emphasizes the relevance of the database over the fitting method. Finally, this study underscores the limitations of root mean square errors and the need for comprehensive testing, advocating the use of multiple MLPs for enhanced certainty, particularly when simulating complex thermodynamic properties that may not be fully captured by simpler tests.},
keywords = {P03, P12},
pubstate = {published},
tppubtype = {article}
}
Falkner, Sebastian; Coretti, Alessandro; Dellago, Christoph
Journal ArticleOpen AccessIn: Physical Review Letters, vol. 132, iss. 12, pp. 128001, 2024.
Abstract | Links | BibTeX | Tags: P12
@article{Falkner2024,
title = {Enhanced Sampling of Configuration and Path Space in a Generalized Ensemble by Shooting Point Exchange},
author = {Sebastian Falkner and Alessandro Coretti and Christoph Dellago},
url = {https://arxiv.org/abs/2302.08757},
doi = {https://doi.org/10.1103/PhysRevLett.132.128001},
year = {2024},
date = {2024-03-18},
urldate = {2024-03-18},
journal = {Physical Review Letters},
volume = {132},
issue = {12},
pages = {128001},
abstract = {The computer simulation of many molecular processes is complicated by long timescales caused by rare transitions between long-lived states. Here, we propose a new approach to simulate such rare events, which combines transition path sampling with enhanced exploration of configuration space. The method relies on exchange moves between configuration and trajectory space, carried out based on a generalized ensemble. This scheme substantially enhances the efficiency of the transition path sampling simulations, particularly for systems with multiple transition channels, and yields information on thermodynamics, kinetics and reaction coordinates of molecular processes without distorting their dynamics. The method is illustrated using the isomerization of proline in the KPTP tetrapeptide.},
keywords = {P12},
pubstate = {published},
tppubtype = {article}
}

Celiberti, Lorenzo; Mosca, Dario Fiore; Allodi, Giuseppe; Pourovskii, Leonid V.; Tassetti, Anna; Forino, Paola Caterina; Cong, Rong; Garcia, Erick; Tran, Phuong M.; Renzi, Roberto De; Woodward, Patrick M.; Mitrović, Vesna F.; Sanna, Samuele; Franchini, Cesare
Spin-orbital Jahn-Teller bipolarons
Journal ArticleOpen AccessIn: Nature Communications, vol. 15, no. 2429, 2024.
Abstract | Links | BibTeX | Tags: P07
@article{Celiberti2024,
title = {Spin-orbital Jahn-Teller bipolarons},
author = {Lorenzo Celiberti and Dario Fiore Mosca and Giuseppe Allodi and Leonid V. Pourovskii and Anna Tassetti and Paola Caterina Forino and Rong Cong and Erick Garcia and Phuong M. Tran and Roberto De Renzi and Patrick M. Woodward and Vesna F. Mitrović and Samuele Sanna and Cesare Franchini},
url = {https://doi.org/10.1038/s41467-024-46621-0},
doi = {10.1038/s41467-024-46621-0},
year = {2024},
date = {2024-03-18},
urldate = {2024-03-18},
journal = {Nature Communications},
volume = {15},
number = {2429},
abstract = {Polarons and spin-orbit (SO) coupling are distinct quantum effects that play a critical role in charge transport and spin-orbitronics. Polarons originate from strong electron-phonon interaction and are ubiquitous in polarizable materials featuring electron localization, in particular 3d transition metal oxides (TMOs). On the other hand, the relativistic coupling between the spin and orbital angular momentum is notable in lattices with heavy atoms and develops in 5d TMOs, where electrons are spatially delocalized. Here we combine ab initio calculations and magnetic measurements to show that these two seemingly mutually exclusive interactions are entangled in the electron-doped SO-coupled Mott insulator Ba_{2}Na_{1−x}Ca_{x}OsO_{6} (0 < x < 1), unveiling the formation of spin-orbital bipolarons. Polaron charge trapping, favoured by the Jahn-Teller lattice activity, converts the Os 5d^{1} spin-orbital J_{eff} = 3/2 levels, characteristic of the parent compound Ba_{2}NaOsO_{6} (BNOO), into a bipolaron 5d^{2} J_{eff} = 2 manifold, leading to the coexistence of different J-effective states in a single-phase material. The gradual increase of bipolarons with increasing doping creates robust in-gap states that prevents the transition to a metal phase even at ultrahigh doping, thus preserving the Mott gap across the entire doping range from d^{1} BNOO to d^{2} Ba_{2}CaOsO_{6} (BCOO).},
keywords = {P07},
pubstate = {published},
tppubtype = {article}
}
Gorfer, Alexander; Abart, Rainer; Dellago, Christoph
Structure and thermodynamics of defects in Na-feldspar from a neural network potential
Journal ArticleSubmittedarXivIn: arXiv, 2024.
Abstract | Links | BibTeX | Tags: P12
@article{Gorfer_2024,
title = {Structure and thermodynamics of defects in Na-feldspar from a neural network potential},
author = {Alexander Gorfer and Rainer Abart and Christoph Dellago},
url = {https://arxiv.org/abs/2402.14640},
year = {2024},
date = {2024-02-22},
urldate = {2024-02-22},
journal = {arXiv},
abstract = {The diffusive phase transformations occurring in feldspar, a common mineral in the crust of the Earth, are essential for reconstructing the thermal histories of magmatic and metamorphic rocks. Due to the long timescales over which these transformations proceed, the mechanism responsible for sodium diffusion and its possible anisotropy has remained a topic of debate. To elucidate this defect-controlled process, we have developed a Neural Network Potential (NNP) trained on first-principle calculations of Na-feldspar (Albite) and its charged defects. This new force field reproduces various experimentally known properties of feldspar, including its lattice parameters, elastic constants as well as heat capacity and DFT-calculated defect formation energies. A new type of dumbbell interstitial defect is found to be most favorable and its free energy of formation at finite temperature is calculated using thermodynamic integration. The necessity of including electrostatic corrections before training an NNP is demonstrated by predicting more consistent defect formation energies.},
keywords = {P12},
pubstate = {published},
tppubtype = {article}
}
Omranpour, Amir; de Hijes, Pablo Montero; Behler, Jörg; Dellago, Christoph
Perspective: Atomistic Simulations of Water and Aqueous Systems with Machine Learning Potentials
Journal ArticleSubmittedarXivIn: arXiv, 2024.
Abstract | Links | BibTeX | Tags: P12
@article{Omranpour_2024,
title = {Perspective: Atomistic Simulations of Water and Aqueous Systems with Machine Learning Potentials},
author = {Amir Omranpour and Pablo Montero de Hijes and Jörg Behler and Christoph Dellago},
url = {https://arxiv.org/abs/2401.17875},
year = {2024},
date = {2024-01-31},
urldate = {2024-01-31},
journal = {arXiv},
abstract = {As the most important solvent, water has been at the center of interest since the advent of computer simulations. While early molecular dynamics and Monte Carlo simulations had to make use of simple model potentials to describe the atomic interactions, accurate ab initio molecular dynamics simulations relying on the first-principles calculation of the energies and forces have opened the way to predictive simulations of aqueous systems. Still, these simulations are very demanding, which prevents the study of complex systems and their properties. Modern machine learning potentials (MLPs) have now reached a mature state, allowing to overcome these limitations by combining the high accuracy of electronic structure calculations with the efficiency of empirical force fields. In this Perspective we give a concise overview about the progress made in the simulation of water and aqueous systems employing MLPs, starting from early work on free molecules and clusters via bulk liquid water to electrolyte solutions and solid-liquid interfaces.},
keywords = {P12},
pubstate = {published},
tppubtype = {article}
}
Wang, Chunlei; Sombut, Panukorn; Puntscher, Lena; Jakub, Zdenek; Meier, Matthias; Pavelec, Jiri; Bliem, Roland; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare; Parkinson, Gareth S.
CO‐Induced Dimer Decay Responsible for Gem‐Dicarbonyl Formation on a Model Single‐Atom Catalyst
Journal ArticleOpen AccessIn PressIn: Angewandte Chemie - International Edition, no. e202317347, 2024, ISSN: 1521-3773.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Wang2024,
title = {CO‐Induced Dimer Decay Responsible for Gem‐Dicarbonyl Formation on a Model Single‐Atom Catalyst},
author = {Chunlei Wang and Panukorn Sombut and Lena Puntscher and Zdenek Jakub and Matthias Meier and Jiri Pavelec and Roland Bliem and Michael Schmid and Ulrike Diebold and Cesare Franchini and Gareth S. Parkinson},
doi = {10.1002/anie.202317347},
issn = {1521-3773},
year = {2024},
date = {2024-01-31},
journal = {Angewandte Chemie - International Edition},
number = {e202317347},
publisher = {Wiley},
abstract = {The ability to coordinate multiple reactants at the same active site is important for the wide-spread applicability of single-atom catalysis. Model catalysts are ideal to investigate the link between active site geometry and reactant binding, because the structure of single-crystal surfaces can be precisely determined, the adsorbates imaged by scanning tunneling microscopy (STM), and direct comparisons made to density functional theory. In this study, we follow the evolution of Rh_{1} adatoms and minority Rh_{2} dimers on Fe_{3}O_{4}(001) during exposure to CO using time-lapse STM at room temperature. CO adsorption at Rh_{1} sites results exclusively in stable Rh_{1}CO monocarbonyls, because the Rh atom adapts its coordination to create a stable pseudo-square planar environment. Rh_{1}(CO)_{2} gem-dicarbonyl species are also observed, but these form exclusively through the breakup of Rh_{2} dimers via an unstable Rh_{2}(CO)_{3} intermediate. Overall, our results illustrate how minority species invisible to area-averaging spectra can play an important role in catalytic systems, and show that the decomposition of dimers or small clusters can be an avenue to produce reactive, metastable configurations in single-atom catalysis.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Birschitzky, Viktor; Sokolovic, Igor; Prezzi, Michael; Palotas, Krisztian; Setvin, Martin; Diebold, Ulrike; Reticcioli, Michele; Franchini, Cesare
Machine Learning Based Prediction of Polaron-Vacancy Patterns on the TiO2(110) Surface
Journal ArticleOpen AccessAccepted ArticlearXivIn: npj Computational Materials, 2024.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Birschitzky_2024a,
title = {Machine Learning Based Prediction of Polaron-Vacancy Patterns on the TiO2(110) Surface},
author = {Viktor Birschitzky and Igor Sokolovic and Michael Prezzi and Krisztian Palotas and Martin Setvin and Ulrike Diebold and Michele Reticcioli and Cesare Franchini},
doi = { https://doi.org/10.48550/arXiv.2401.12042},
year = {2024},
date = {2024-01-22},
journal = {npj Computational Materials},
abstract = {The multifaceted physics of oxides is shaped by their composition and the presence of defects, which are often accompanied by the formation of polarons. The simultaneous presence of polarons and defects, and their complex interactions, pose challenges for first-principles simulations and experimental techniques. In this study, we leverage machine learning and a first-principles database to analyze the distribution of surface oxygen vacancies (VO) and induced small polarons on rutile TiO2(110), effectively disentangling the interactions between polarons and defects. By combining neural-network supervised learning and simulated annealing, we elucidate the inhomogeneous VO distribution observed in scanning probe microscopy (SPM). Our innovative approach allows us to understand and predict defective surface patterns at previously inaccessible length scales, identifying the specific role of individual types of defects. Specifically, surface-polaron-stabilizing VO-configurations are identified, which could have consequences for surface reactivity.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
2023
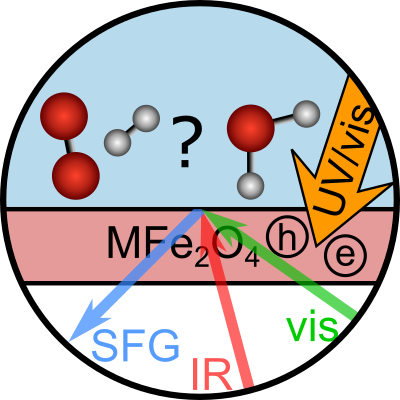
Backus, Ellen H. G.; Hosseinpour, Saman; Ramanan, Charusheela; Sun, Shumei; Schlegel, Simon J.; Zelenka, Moritz; Jia, Xiaoyu; Gebhard, Maximilian; Devi, Anjana; Wang, Hai I.; Bonn, Mischa
Journal ArticleOpen AccessIn: Angewandte Chemie - International Edition, no. e202312123, 2023, ISSN: 1521-3773.
Abstract | Links | BibTeX | Tags: P11
@article{Backus2024,
title = {Ultrafast Surface‐Specific Spectroscopy of Water at a Photoexcited TiO2 Model Water‐Splitting Photocatalyst},
author = {Ellen H. G. Backus and Saman Hosseinpour and Charusheela Ramanan and Shumei Sun and Simon J. Schlegel and Moritz Zelenka and Xiaoyu Jia and Maximilian Gebhard and Anjana Devi and Hai I. Wang and Mischa Bonn},
doi = {10.1002/anie.202312123},
issn = {1521-3773},
year = {2023},
date = {2023-11-27},
urldate = {2023-11-27},
journal = {Angewandte Chemie - International Edition},
number = {e202312123},
publisher = {Wiley},
abstract = {A critical step in photocatalytic water dissociation is the hole-mediated oxidation reaction. Molecular-level insights into the mechanism of this complex reaction under realistic conditions with high temporal resolution are highly desirable. Here, we use femtosecond time-resolved, surface-specific vibrational sum frequency generation spectroscopy to study the photo-induced reaction directly at the interface of the photocatalyst TiO_{2} in contact with liquid water at room temperature. Thanks to the inherent surface specificity of the spectroscopic method, we can follow the reaction of solely the interfacial water molecules directly at the interface at timescales on which the reaction takes place. Following the generation of holes at the surface immediately after photoexcitation of the catalyst with UV light, water dissociation occurs on a sub-20 ps timescale. The reaction mechanism is similar at pH 3 and 11. In both cases, we observe the conversion of H_{2}O into Ti−OH groups and the deprotonation of pre-existing Ti−OH groups. This study provides unique experimental insights into the early steps of the photo-induced dissociation processes at the photocatalyst-water interface, relevant to the design of improved photocatalysts.},
keywords = {P11},
pubstate = {published},
tppubtype = {article}
}