
A collaborative article of our colleagues of P02 was accepted into Angewandte Chemie and additionally ranked in the top 10 % of articles.

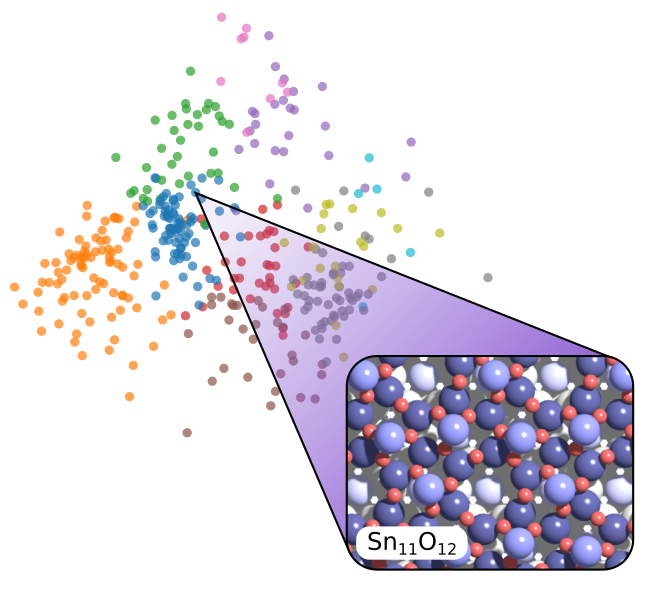
An essential step towards optimal search algorithms for surface structures was recently achieved by a team of authors, including the TACO scientists Ulrike Diebold and Michael Schmid, as well as the TACO Scientific Advisory Board member Bjørk Hammer. The team applied an evolutionary algorithm, enhanced by machine-learning methods, to the structure of an ultrathin tin oxide film on a platinum-tin alloy – the Pt3Sn(111) surface, to be precise. These materials are important for, e.g., electrocatalytic processes and serve as model systems for realistic materials. However, the oxide surface and its interface to the alloy below remained poorly understood at the atomic level. In the past, the structure proved to be too complex to be directly deduced from experimental data by human chemical intuition alone. The computational cost of global optimisation methods at the level of density functional theory (DFT), on the other hand, has been prohibitively high. Here, machine-learned approximations come to the rescue, which have achieved orders-of-magnitude reductions in computation times. They can reach reliable results based on only a limited set of detailed but costly DFT calculations. The specialty of the evolutionary code developed for the present work is that it retains the entire search history for a global energy minimum and thus uses all available data. This design makes it both efficient and reliable and avoids premature convergence to a non-global energy minimum.
The authors applied only very few general assumptions on the composition and structure of the surface beforehand. Eventually, it turned out that the surface tin oxide has a Sn11O12 composition in a (4×4) unit cell, with three tin atoms per unit cell protruding from the surface – a highly non-trivial structure. Nevertheless, the properties of the identified optimal, lowest-energy structure predicted by the model agree very well with the available experimental data. The structure was verified experimentally by scanning probe microscopy images with atomic resolution and surface X-ray diffraction. Thus, the results are highly promising as novel way to overcome previous limitations in deducing surface structures both from experimental data and from DFT simulations at acceptable computational costs.
The work by Lindsay R. Merte et al. is presented in an article that was recently accepted for publication in the journal Angewandte Chemie. Adding to this achievement, the article’s referees evaluated its results to be within the top 10 % in the journal, meaning that this research article is considered “highly important”.
Abstract:
Determination of the atomic structure of solid surfaces typically depends on comparison of measured properties with simulations based on hypothesized structural models. For simple structures, the models may be guessed, but for more complex structures there is a need for reliable theory-based search algorithms. So far, such methods have been limited by the combinatorial complexity and computational expense of sufficiently accurate energy estimation for surfaces. However, the introduction of machine learning methods has the potential to change this radically. Here, we demonstrate how an evolutionary algorithm, utilizing machine learning for accelerated energy estimation and diverse population generation, can be used to solve an unknown surface structure—the (4×4) surface oxide on Pt3Sn(111)–based on limited experimental input. The algorithm is efficient and robust, and should be broadly applicable in surface studies, where it can replace manual, intuition based model generation.
The full article can be found here.
Authors:
Lindsay R. Merte, Malthe Kjær Bisbo, Igor Sokolović, Martin Setvín, Benjamin Hagman, Mikhail Shipilin, Michael Schmid, Ulrike Diebold, Edvin Lundgren, and Bjørk Hammer
Subprojects:
P02 – Surface structure and reactivity of multi-component oxides at the atomic scale
