
Bayesian regression for
multi-level machine-learned potentials
Subproject P03
The first-principles description of the properties of multi-component metal oxides is an exceedingly challenging problem. The reasons are that the configurational space grows exponentially with the number of species and standard Density Functional Theory (DFT) is often not accurate enough. The long-term objective of P03 is to accelerate first-principles calculations by developing machine-learning approaches for the description of the interatomic forces, Born effective charges, and other tensorial properties of multivalent oxides. The project will rely on kernel-based methods and Bayesian inference to implement fully automatic “on-the-fly” learning.
In the first project period, we will develop machine-learned force fields (MLFF) for DFT and DFT+U, whereby the number of components in the FF will be gradually increased. A concise framework for learning tensorial properties will be implemented. We will use this to simulate infrared spectra of oxide materials, which can be readily compared to the finite-temperature spectra measured by the experimental groups.
The difference between DFT and hybrid functionals will be machine-learned to go beyond semi-local functionals (Delta-learning). The long-term perspective is to extend this approach to highly accurate beyond-DFT methods, such as the random phase approximation and quantum chemistry (coupled cluster) methods. Although kernel-based methods are exceedingly accurate, they are often less efficient than NN. We will collaborate with other projects to recast the on-the-fly trained FF into NN potentials to address this issue.


Georg Kresse
PI
Expertise
The main research efforts of the group are directed towards the development of quantum-mechanical tools for atomic-scale simulations of properties and processes in materials and the application of these methodologies to key areas of condensed matter physics and materials research. An important pillar of the research is the Vienna Ab initio Simulation Package (VASP), a general-purpose ab initio code for solving the many-electron Schrödinger equation. The code is among the world leaders in its field, with more than 3500 licensees worldwide. We have expertise with simulations for a vast number of properties using many different techniques:
- Density functional theory (DFT), including spin and non-collinear DFT
- Linear response theory to calculate phonons and dielectric properties
- Hartree-Fock techniques and many flavors of hybrid functionals
- Many-body perturbation theory, including GW and Bethe-Salpeter
- Wavefunction-based correlated methods (Møller-Plesset perturbation theory)
- Surface science, including growth and oxide formation
- Simulation of nanostructures
- Semiconductor physics: charge trapping, polarons
- Electronic excitations
- Defect energies in extended systems
For TACO, we will adapt our machine-learning techniques to tensorial properties and correlated wavefunction techniques. These techniques are directly integrated into VASP and allow to accelerate finite-temperature simulations by many orders of magnitudes.
Team

Georg Kresse
PI

Bernhard Schmiedmayer
PhD Student

Carolin Faller PhD Student, Student Representative 22–24
Former Members

Carla Verdi
co-PI

Peitao Liu
PostDoc
Publications
2020
Yang, Jingxia; Ding, Huihui; Wang, Jinjie; Yigit, Nevzat; Xu, Jingli; Rupprechter, Günther; Zhang, Min; Li, Zhiquan
Energy-Guided Shape Control Towards Highly Active CeO2
Journal ArticleIn: Topics in Catalysis, vol. 63, no. 19-20, pp. 1743–1753, 2020.
Abstract | Links | BibTeX | Tags: P08, pre-TACO
@article{Yang2020,
title = {Energy-Guided Shape Control Towards Highly Active CeO_{2}},
author = {Jingxia Yang and Huihui Ding and Jinjie Wang and Nevzat Yigit and Jingli Xu and Günther Rupprechter and Min Zhang and Zhiquan Li},
doi = {10.1007/s11244-020-01357-1},
year = {2020},
date = {2020-08-18},
journal = {Topics in Catalysis},
volume = {63},
number = {19-20},
pages = {1743--1753},
publisher = {Springer Science and Business Media LLC},
abstract = {The shape of nanosized CeO_{2}, obtained via polyvinylpyrrolidone (PVP) micelles, was controlled by microwave (MW)-aided synthesis combined with different combinations of energy input/transfer, including ultrasound (US), ultraviolet (UV) and pressure (P). Whereas ceria nanoflakes resulted from standard solvothermal synthesis, CeO_{2} nanoparticles were obtained from MW, MW + US and MW + UV. New CeO_{2} “nanospindles” (with aspect ratio of 2) resulted from MW + US + UV, and nanorods (with aspect ratio of 11) emerged from MW + P. All ceria morphologies, even nanospindles and nanorods, were mesoporous agglomerates of small CeO_{2} nanocrystals (6–8 nm size), but they still exhibited different specific surface area (SSA) and Ce^{3+}/Ce^{4+} ratio. Underlying reasons of how the different synthesis routes affect the ceria morphology are discussed. Among the six types, CeO_{2} nanorods (MW + P) exhibited the highest SSA (196 m^{2} g^{−1}) and the most surface defects (Ce^{3+}: 26.4%), resulting in excellent catalytic performance in imine synthesis and CO oxidation.},
keywords = {P08, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Jinnouchi, Ryosuke; Miwa, Kazutoshi; Karsai, Ferenc; Kresse, Georg; Asahi, Ryoji
On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations
Journal ArticleIn: The Journal of Physical Chemistry Letters, vol. 11, no. 17, pp. 6946–6955, 2020.
Abstract | Links | BibTeX | Tags: P03, pre-TACO
@article{Jinnouchi2020,
title = {On-the-Fly Active Learning of Interatomic Potentials for Large-Scale Atomistic Simulations},
author = {Ryosuke Jinnouchi and Kazutoshi Miwa and Ferenc Karsai and Georg Kresse and Ryoji Asahi},
doi = {10.1021/acs.jpclett.0c01061},
year = {2020},
date = {2020-07-31},
journal = {The Journal of Physical Chemistry Letters},
volume = {11},
number = {17},
pages = {6946--6955},
publisher = {American Chemical Society (ACS)},
abstract = {The on-the-fly generation of machine-learning force fields by active-learning schemes attracts a great deal of attention in the community of atomistic simulations. The algorithms allow the machine to self-learn an interatomic potential and construct machine-learned models on the fly during simulations. State-of-the-art query strategies allow the machine to judge whether new structures are out of the training data set or not. Only when the machine judges the necessity of updating the data set with the new structures are first-principles calculations carried out. Otherwise, the yet available machine-learned model is used to update the atomic positions. In this manner, most of the first-principles calculations are bypassed during training, and overall, simulations are accelerated by several orders of magnitude while retaining almost first-principles accuracy. In this Perspective, after describing essential components of the active-learning algorithms, we demonstrate the power of the schemes by presenting recent applications.},
keywords = {P03, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Grumelli, Doris; Wiegmann, Tim; Barja, Sara; Reikowski, Finn; Maroun, Fouad; Allongue, Philippe; Balajka, Jan; Parkinson, Gareth S.; Diebold, Ulrike; Kern, Klaus; Magnussen, Olaf M
Electrochemical Stability of the Reconstructed Fe3O4(001) Surface
Journal ArticleIn: Angewandte Chemie - International Edition, vol. 59, no. 49, pp. 21904–21908, 2020.
Abstract | Links | BibTeX | Tags: P02, P04, pre-TACO
@article{Grumelli2020,
title = {Electrochemical Stability of the Reconstructed Fe_{3}O_{4}(001) Surface},
author = {Doris Grumelli and Tim Wiegmann and Sara Barja and Finn Reikowski and Fouad Maroun and Philippe Allongue and Jan Balajka and Gareth S. Parkinson and Ulrike Diebold and Klaus Kern and Olaf M Magnussen},
doi = {10.1002/anie.202008785},
year = {2020},
date = {2020-07-29},
urldate = {2020-07-29},
journal = {Angewandte Chemie - International Edition},
volume = {59},
number = {49},
pages = {21904--21908},
publisher = {Wiley},
abstract = {Establishing the atomic-scale structure of metal-oxide surfaces during electrochemical reactions is a key step to modeling this important class of electrocatalysts. Here, we demonstrate that the characteristic (√2×√2)R45° surface reconstruction formed on (001)-oriented magnetite single crystals is maintained after immersion in 0.1 M NaOH at 0.20 V vs. Ag/AgCl and we investigate its dependence on the electrode potential. We follow the evolution of the surface using in situ and operando surface X-ray diffraction from the onset of hydrogen evolution, to potentials deep in the oxygen evolution reaction (OER) regime. The reconstruction remains stable for hours between −0.20 and 0.60 V and, surprisingly, is still present at anodic current densities of up to 10 mA cm^{−2} and strongly affects the OER kinetics. We attribute this to a stabilization of the Fe_{3}O_{4} bulk by the reconstructed surface. At more negative potentials, a gradual and largely irreversible lifting of the reconstruction is observed due to the onset of oxide reduction.},
keywords = {P02, P04, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Suchorski, Yuri; Rupprechter, Günther
Catalysis by Imaging: From Meso- to Nano-scale
Journal ArticleOpen AccessIn: Topics in Catalysis, vol. 63, no. 15-18, pp. 1532–1544, 2020.
Abstract | Links | BibTeX | Tags: P08, pre-TACO
@article{Suchorski2020,
title = {Catalysis by Imaging: From Meso- to Nano-scale},
author = {Yuri Suchorski and Günther Rupprechter},
doi = {10.1007/s11244-020-01302-2},
year = {2020},
date = {2020-07-02},
urldate = {2020-07-02},
journal = {Topics in Catalysis},
volume = {63},
number = {15-18},
pages = {1532--1544},
publisher = {Springer Science and Business Media LLC},
abstract = {In-situ imaging of catalytic reactions has provided insights into reaction front propagation, pattern formation and other spatio-temporal effects for decades. Most recently, analysis of the local image intensity opened a way towards evaluation of local reaction kinetics. Herein, our recent studies of catalytic CO oxidation on Pt(hkl) and Rh(hkl) via the kinetics by imaging approach, both on the meso- and nano-scale, are reviewed. Polycrystalline Pt and Rh foils and nanotips were used as µm- and nm-sized surface structure libraries as model systems for reactions in the 10^{–5}–10^{–6} mbar pressure range. Isobaric light-off and isothermal kinetic transitions were visualized in-situ at µm-resolution by photoemission electron microscopy (PEEM), and at nm-resolution by field emission microscopy (FEM) and field ion microscopy (FIM). The local reaction kinetics of individual Pt(hkl) and Rh(hkl) domains and nanofacets of Pt and Rh nanotips were deduced from the local image intensity analysis. This revealed the structure-sensitivity of CO oxidation, both in the light-off and in the kinetic bistability: for different low-index Pt surfaces, differences of up to 60 K in the critical light-off temperatures and remarkable differences in the bistability ranges of differently oriented stepped Rh surfaces were observed. To prove the spatial coherence of light-off on nanotips, proper orthogonal decomposition (POD) as a spatial correlation analysis was applied to the FIM video-data. The influence of particular configurations of steps and kinks on kinetic transitions were analysed by using the average nearest neighbour number as a common descriptor. Perspectives of nanosized surface structure libraries for future model studies are discussed.},
keywords = {P08, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Backus, Ellen H. G.; Schaefer, Jan; Bonn, Mischa
Probing the Mineral–Water Interface with Nonlinear Optical Spectroscopy
Journal ArticleOpen AccessIn: Angewandte Chemie - International Edition, vol. 60, no. 19, pp. 10482–10501, 2020.
Abstract | Links | BibTeX | Tags: P11, pre-TACO
@article{Backus2020,
title = {Probing the Mineral–Water Interface with Nonlinear Optical Spectroscopy},
author = {Ellen H. G. Backus and Jan Schaefer and Mischa Bonn},
doi = {10.1002/anie.202003085},
year = {2020},
date = {2020-06-19},
urldate = {2020-06-19},
journal = {Angewandte Chemie - International Edition},
volume = {60},
number = {19},
pages = {10482--10501},
publisher = {Wiley},
abstract = {The interaction between minerals and water is manifold and complex: the mineral surface can be (de)protonated by water, thereby changing its charge; mineral ions dissolved into the aqueous phase screen the surface charges. Both factors affect the interaction with water. Intrinsically molecular-level processes and interactions govern macroscopic phenomena, such as flow-induced dissolution, wetting, and charging. This realization is increasingly prompting molecular-level studies of mineral–water interfaces. Here, we provide an overview of recent developments in surface-specific nonlinear spectroscopy techniques such as sum frequency and second harmonic generation (SFG/SHG), which can provide information about the molecular arrangement of the first few layers of water molecules at the mineral surface. The results illustrate the subtleties of both chemical and physical interactions between water and the mineral as well as the critical role of mineral dissolution and other ions in solution for determining those interactions.},
keywords = {P11, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Pollitt, Stephan; Truttmann, Vera; Haunold, Thomas; Garcia, Clara; Olszewski, Wojciech; Llorca, Jordi; é, Noelia Barrab; Rupprechter, Günther
Journal ArticleOpen AccessIn: ACS Catalysis, vol. 10, no. 11, pp. 6144–6148, 2020.
Abstract | Links | BibTeX | Tags: P08, pre-TACO
@article{Pollitt2020,
title = {The Dynamic Structure of Au_{38}(SR)_{24} Nanoclusters Supported on CeO_{2} upon Pretreatment and CO Oxidation},
author = {Stephan Pollitt and Vera Truttmann and Thomas Haunold and Clara Garcia and Wojciech Olszewski and Jordi Llorca and Noelia Barrab é and Günther Rupprechter},
doi = {10.1021/acscatal.0c01621},
year = {2020},
date = {2020-05-08},
urldate = {2020-05-08},
journal = {ACS Catalysis},
volume = {10},
number = {11},
pages = {6144--6148},
publisher = {American Chemical Society (ACS)},
abstract = {Atomically precise thiolate protected Au nanoclusters Au38}(SC_{2}H_{4}Ph)_{24} on CeO_{2} were used for in-situ (operando) extended X-ray absorption fine structure/diffuse reflectance infrared fourier transform spectroscopy and ex situ scanning transmission electron microscopy–high-angle annular dark-field imaging/X-ray photoelectron spectroscopy studies monitoring cluster structure changes induced by activation (ligand removal) and CO oxidation. Oxidative pretreatment at 150 °C “collapsed” the clusters’ ligand shell, oxidizing the hydrocarbon backbone, but the S remaining on Au acted as poison. Oxidation at 250 °C produced bare Au surfaces by removing S which migrated to the support (forming Au^{+}-S), leading to highest activity. During reaction, structural changes occurred via CO-induced Au and O-induced S migration to the support. The results reveal the dynamics of nanocluster catalysts and the underlying cluster chemistry.},
keywords = {P08, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Lesnicki, Dominika; Zhang, Zhen; Bonn, Mischa; Sulpizi, Marialore; Backus, Ellen H. G.
Journal ArticleOpen AccessIn: Angewandte Chemie - International Edition, vol. 59, no. 31, pp. 13116–13121, 2020.
Abstract | Links | BibTeX | Tags: P11, pre-TACO
@article{Lesnicki2020,
title = {Surface Charges at the CaF_{2}/Water Interface Allow Very Fast Intermolecular Vibrational-Energy Transfer},
author = {Dominika Lesnicki and Zhen Zhang and Mischa Bonn and Marialore Sulpizi and Ellen H. G. Backus},
doi = {10.1002/anie.202004686},
year = {2020},
date = {2020-04-02},
urldate = {2020-04-02},
journal = {Angewandte Chemie - International Edition},
volume = {59},
number = {31},
pages = {13116--13121},
publisher = {Wiley},
abstract = {We investigate the dynamics of water in contact with solid calcium fluoride, where at low pH, localized charges can develop upon fluorite dissolution. We use 2D surface-specific vibrational spectroscopy to quantify the heterogeneity of the interfacial water (D_{2}O) molecules and provide information about the sub-picosecond vibrational-energy-relaxation dynamics at the buried solid/liquid interface. We find that strongly H-bonded OD groups, with a vibrational frequency below 2500 cm^{−1}, display very rapid spectral diffusion and vibrational relaxation; for weakly H-bonded OD groups, above 2500 cm^{−1}, the dynamics slows down substantially. Atomistic simulations based on electronic-structure theory reveal the molecular origin of energy transport through the local H-bond network. We conclude that strongly oriented H-bonded water molecules in the adsorbed layer, whose orientation is pinned by the localized charge defects, can exchange vibrational energy very rapidly due to the strong collective dipole, compensating for a partially missing solvation shell.},
keywords = {P11, pre-TACO},
pubstate = {published},
tppubtype = {article}
}

Lindenthal, Lorenz; Rameshan, Raffael; Summerer, Harald; Ruh, Thomas; Popovic, Janko; Nenning, Andreas; Löffler, Stefan; Opitz, Alexander Karl; Blaha, Peter; Rameshan, Christoph
Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution
Journal ArticleOpen AccessIn: Catalysts, vol. 10, no. 3, pp. 268, 2020.
Abstract | Links | BibTeX | Tags: P10, pre-TACO
@article{Lindenthal2020,
title = {Modifying the Surface Structure of Perovskite-Based Catalysts by Nanoparticle Exsolution},
author = {Lorenz Lindenthal and Raffael Rameshan and Harald Summerer and Thomas Ruh and Janko Popovic and Andreas Nenning and Stefan Löffler and Alexander Karl Opitz and Peter Blaha and Christoph Rameshan},
doi = {10.3390/catal10030268},
year = {2020},
date = {2020-03-01},
urldate = {2020-03-01},
journal = {Catalysts},
volume = {10},
number = {3},
pages = {268},
publisher = {MDPI AG},
abstract = {In heterogeneous catalysis, surfaces decorated with uniformly dispersed, catalytically-active (nano)particles are a key requirement for excellent performance. Beside standard catalyst preparation routines—with limitations in controlling catalyst surface structure (i.e., particle size distribution or dispersion)—we present here a novel time efficient route to precisely tailor catalyst surface morphology and composition of perovskites. Perovskite-type oxides of nominal composition ABO3 with transition metal cations on the B-site can exsolve the B-site transition metal upon controlled reduction. In this exsolution process, the transition metal emerges from the oxide lattice and migrates to the surface where it forms catalytically active nanoparticles. Doping the B-site with reducible and catalytically highly active elements, offers the opportunity of tailoring properties of exsolution catalysts. Here, we present the synthesis of two novel perovskite catalysts Nd_{0.6}Ca_{0.4}FeO_{3-δ} and Nd_{0.6}Ca_{0.4}Fe_{0.9}Co_{0.1}O_{3-δ} with characterisation by (in situ) XRD, SEM/TEM and XPS, supported by theory (DFT+U). Fe nanoparticle formation was observed for Nd_{0.6}Ca_{0.4}FeO_{3-δ}. In comparison, B site cobalt doping leads, already at lower reduction temperatures, to formation of finely dispersed Co nanoparticles on the surface. These novel perovskite-type catalysts are highly promising for applications in chemical energy conversion. First measurements revealed that exsolved Co nanoparticles significantly improve the catalytic activity for CO_{2} activation via reverse water gas shift reaction. },
keywords = {P10, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
2019
Michl, Jakob; Sega, Marcello; Dellago, Christoph
Phase stability of the ice XVII-based CO2 chiral hydrate from molecular dynamics simulations
Journal ArticleOpen AccessIn: The Journal of Chemical Physics, vol. 151, no. 10, pp. 104502, 2019.
Abstract | Links | BibTeX | Tags: P12, pre-TACO
@article{Michl2019,
title = {Phase stability of the ice XVII-based CO_{2} chiral hydrate from molecular dynamics simulations},
author = {Jakob Michl and Marcello Sega and Christoph Dellago},
doi = {10.1063/1.5116540},
year = {2019},
date = {2019-09-12},
urldate = {2019-09-12},
journal = {The Journal of Chemical Physics},
volume = {151},
number = {10},
pages = {104502},
publisher = {AIP Publishing},
abstract = {We computed the phase diagram of CO_{2} hydrates at high pressure (HP), from 0.3 to 20 kbar, by means of molecular dynamics simulations. The two CO_{2} hydrates known to occur in this pressure range are the cubic structure I (sI) clathrate and the HP hydrate, whose water framework is the recently discovered ice XVII. We investigated the stability of both hydrates upon heating (melting) as well as the phase changes upon compression. The CO_{2}-filled ice XVII is found to be more stable than the sI clathrate and than the mixture of ice VI and dry ice at pressure values ranging from 6 to 18 kbar and in a wide temperature range, although a phenomenological correction suggests that the stability should more realistically range from 6.5 to 13.5 kbar. Our simulation results support the current hypothesis that the HP hydrate is stable at temperatures above the melting curve of ice VI.},
keywords = {P12, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
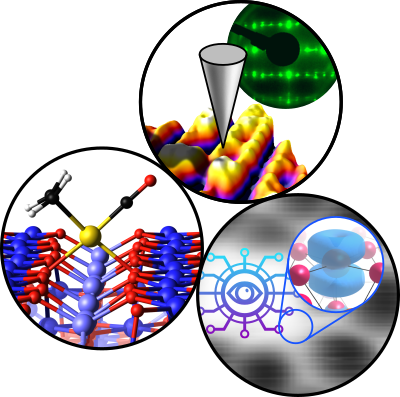
Jakub, Zdenek; Hulva, Jan; Meier, Matthias; Bliem, Roland; Kraushofer, Florian; Setvin, Martin; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare; Parkinson, Gareth S.
Local Structure and Coordination Define Adsorption in a Model Ir1/Fe3O4 Single-Atom Catalyst
Journal ArticleOpen AccessIn: Angewandte Chemie - International Edition, vol. 58, no. 39, pp. 13961–13968, 2019.
Abstract | Links | BibTeX | Tags: P02, P04, P07, pre-TACO
@article{Jakub2019,
title = {Local Structure and Coordination Define Adsorption in a Model Ir_{1}/Fe_{3}O_{4} Single-Atom Catalyst},
author = {Zdenek Jakub and Jan Hulva and Matthias Meier and Roland Bliem and Florian Kraushofer and Martin Setvin and Michael Schmid and Ulrike Diebold and Cesare Franchini and Gareth S. Parkinson},
doi = {10.1002/anie.201907536},
year = {2019},
date = {2019-07-24},
urldate = {2019-07-24},
journal = {Angewandte Chemie - International Edition},
volume = {58},
number = {39},
pages = {13961--13968},
publisher = {Wiley},
abstract = {Single-atom catalysts (SACs) bridge homo- and heterogeneous catalysis because the active site is a metal atom coordinated to surface ligands. The local binding environment of the atom should thus strongly influence how reactants adsorb. Now, atomically resolved scanning-probe microscopy, X-ray photoelectron spectroscopy, temperature-programmed desorption, and DFT are used to study how CO binds at different Ir_{1} sites on a precisely defined Fe_{3}O_{4}(001) support. The two- and five-fold-coordinated Ir adatoms bind CO more strongly than metallic Ir, and adopt structures consistent with square-planar Ir^{I} and octahedral Ir^{III} complexes, respectively. Ir incorporates into the subsurface already at 450 K, becoming inactive for adsorption. Above 900 K, the Ir adatoms agglomerate to form nanoparticles encapsulated by iron oxide. These results demonstrate the link between SAC systems and coordination complexes, and that incorporation into the support is an important deactivation mechanism.},
keywords = {P02, P04, P07, pre-TACO},
pubstate = {published},
tppubtype = {article}
}