
Surface structure and reactivity of
multi-component oxides at the atomic scale
Subproject P02
Multi-component metal oxides exhibit a plethora of stoichiometry-dependent structural phases at the surface, even if the composition of the bulk is kept the same. The long-term objective of P02 is to unravel the relationship between surface electronic and geometric structure and reactivity, to ultimately tune these materials for energy-related reactions such as the ORR. The project applies the surface science approach. We will grow well-defined, epitaxial perovskite thin films of LSFO and LSMO in a UHV-based PLD/surface science apparatus under tight control of the surface stoichiometry in the first project period. We will determine the coordinates of surface atoms quantitatively using LEED-IV in close collaboration with theoretical groups.
Theoretical models will also help with interpreting atomically-resolved ncAFM/STM images. These images give direct insights into the behavior of polarons in these complex materials and show how adsorbates such as O2, H2O, CO, and CO2 interact with electronic and structural defects. XPS, TPD, and FTIR of these well-defined systems will deliver desorption energies, vibrational frequencies, and spectral fingerprints. These experimental data on well-defined systems will build a bridge when tested under ‘realistic’ environments at high pressure/temperature and in aqueous solutions. They will also serve to validate ML-based theory approaches.


Ulrike Diebold
PI
Expertise
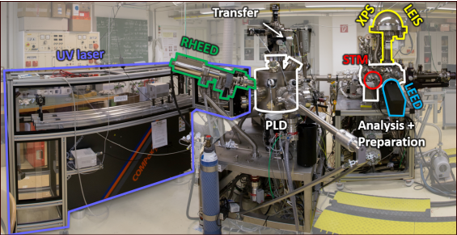
Our expertise is experimental surface science. We operate a total of seven ultrahigh-vacuum (UHV) chambers, which contain virtually all main experimental surface science techniques, as well as an (electro-)chemistry lab.
All chambers are equipped with facilities for sample preparation (sputtering/annealing/gas dosing), as well as various growth techniques (e-beam evaporators, Knudsen cells, UHV-compatible sputter deposition, pulsed laser deposition (PLD)).
Analysis techniques used in our research include:
- Scanning Tunneling Microscopy (STM) (in UHV 4K – 300 K, electrochemical STM)
- Atomic Force Microscopy (AFM): UHV-based (q+ sensor) and in the ambient (cantilever-based)
- Low-Energy Electron Diffraction (LEED)
- Reflection High Energy Diffraction (RHEED)
- X-ray Photoelectron Spectroscopy (XPS)
- Ultraviolet Photoelectron Spectroscopy (UPS)
- Auger Electron Spectroscopy (AES)
- Low-energy He+ ion scattering (LEIS)
- Thermal Programmed Desorption Spectroscopy (TPD)
Of particular use for TACO is our combined Pulsed-Laser Deposition/Surface Science setup shown on the right-hand side. It allows the growth of multi-component metal oxides and the investigations of surface properties within the same UHV setup.
Team

Ulrike Diebold
PI

Michele Riva
co-PI

Michael Schmid
co-PI

Florian Dörr
PhD Student

Alexander Imre
PhD Student, Student Representative 23–24

Paul Haidegger
Master Student

Marie Kienzer
Master Student
Associates

Sarah Tobisch
PhD Student
Former Members

Michael Brunthaler
Master Student

Erik Rheinfrank
PhD-Student

Christoph Schattauer
PostDoc
Publications
2024
Redondo, Jesus; Reticcioli, Michele; Gabriel, Vit; Wrana, Dominik; Ellinger, Florian; Riva, Michele; Franceschi, Giada; Rheinfrank, Erik; Sokolovic, Igor; Jakub, Zdenek; Kraushofer, Florian; Alexander, Aji; Patera, Laerte L.; Repp, Jascha; Schmid, Michael; Diebold, Ulrike; Parkinson, Gareth S.; Franchini, Cesare; Kocan, Pavel; Setvin, Martin
Real-space investigation of polarons in hematite Fe2O3
Journal ArticleOpen AccessIn: Science Advances, vol. 10, iss. 44, pp. eadp7833, 2024.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Redondo2024,
title = {Real-space investigation of polarons in hematite Fe_{2}O_{3}},
author = {Jesus Redondo and Michele Reticcioli and Vit Gabriel and Dominik Wrana and Florian Ellinger and Michele Riva and Giada Franceschi and Erik Rheinfrank and Igor Sokolovic and Zdenek Jakub and Florian Kraushofer and Aji Alexander and Laerte L. Patera and Jascha Repp and Michael Schmid and Ulrike Diebold and Gareth S. Parkinson and Cesare Franchini and Pavel Kocan and Martin Setvin},
url = {https://arxiv.org/abs/2303.17945
https://www.science.org/doi/10.1126/sciadv.adp7833},
year = {2024},
date = {2024-11-01},
urldate = {2024-09-27},
journal = {Science Advances},
volume = {10},
issue = {44},
pages = {eadp7833},
abstract = {In polarizable materials, electronic charge carriers interact with the surrounding ions, leading to quasiparticle behaviour. The resulting polarons play a central role in many materials properties including electrical transport, optical properties, surface reactivity and magnetoresistance, and polaron properties are typically investigated indirectly through such macroscopic characteristics. Here, noncontact atomic force microscopy (nc-AFM) is used to directly image polarons in Fe_{2}O_{3} at the single quasiparticle limit. A combination of Kelvin probe force microscopy (KPFM) and kinetic Monte Carlo (KMC) simulations shows that Ti doping dramatically enhances the mobility of electron polarons, and density functional theory (DFT) calculations indicate that a metallic transition state is responsible for the enhancement. In contrast, hole polarons are significantly less mobile and their hopping is hampered further by the introduction of trapping centres.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Ryan, Paul T. P.; Sombut, Panukorn; Rafsanjani-Abbasi, Ali; Wang, Chunlei; Eratam, Fulden; Goto, Francesco; Diebold, Ulrike; Meier, Matthias; Duncan, David A.; Parkinson, Gareth S.
Journal ArticleOpen AccessIn: The Journal of Physical Chemistry C, vol. 128, iss. 40, pp. 16977–16985, 2024.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Ryan_2024a,
title = {Quantitative Measurement of Cooperative Binding in Partially Dissociated Water Dimers at the Hematite “R-Cut” Surface},
author = {Paul T. P. Ryan and Panukorn Sombut and Ali Rafsanjani-Abbasi and Chunlei Wang and Fulden Eratam and Francesco Goto and Ulrike Diebold and Matthias Meier and David A. Duncan and Gareth S. Parkinson},
url = {https://arxiv.org/abs/2406.18264
https://pubs.acs.org/doi/10.1021/acs.jpcc.4c04537},
year = {2024},
date = {2024-09-30},
urldate = {2024-09-30},
journal = {The Journal of Physical Chemistry C},
volume = {128},
issue = {40},
pages = {16977–16985},
abstract = {Water–solid interfaces pervade the natural environment and modern technology. On some surfaces, water–water interactions induce the formation of partially dissociated interfacial layers; understanding why is important to model processes in catalysis or mineralogy. The complexity of the partially dissociated structures often makes it difficult to probe them quantitatively. Here, we utilize normal incidence X-ray standing waves (NIXSW) to study the structure of partially dissociated water dimers (H_{2}O–OH) at the α-Fe_{2}O_{3}(012) surface (also called the (11̅02) or “R-cut” surface): a system simple enough to be tractable yet complex enough to capture the essential physics. We find the H_{2}O and terminal OH groups to be the same height above the surface within experimental error (1.45 ± 0.04 and 1.47 ± 0.02 Å, respectively), in line with DFT-based calculations that predict comparable Fe–O bond lengths for both water and OH species. This result is understood in the context of cooperative binding, where the formation of the H-bond between adsorbed H_{2}O and OH induces the H_{2}O to bind more strongly and the OH to bind more weakly compared to when these species are isolated on the surface. The surface OH formed by the liberated proton is found to be in plane with a bulk truncated (012) surface (−0.01 ± 0.02 Å). DFT calculations based on various functionals correctly model the cooperative effect but overestimate the water–surface interaction.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Rafsanjani-Abbasi, Ali; Buchner, Florian; Lewis, Faith J.; Puntscher, Lena; Kraushofer, Florian; Sombut, Panukorn; Eder, Moritz; Pavelec, Jiří; Rheinfrank, Erik; Franceschi, Giada; Birschitzky, Viktor; Riva, Michele; Franchini, Cesare; Schmid, Michael; Diebold, Ulrike; Meier, Matthias; Madsen, Georg K. H.; Parkinson, Gareth S.
Digging Its Own Site: Linear Coordination Stabilizes a Pt1/Fe2O3 Single-Atom Catalyst
Journal ArticleOpen AccessIn: ACS Nano, vol. 18, iss. 39, pp. 26920–26927, 2024.
Abstract | Links | BibTeX | Tags: P02, P04, P07, P09
@article{Rafsanjani_2024a,
title = {Digging Its Own Site: Linear Coordination Stabilizes a Pt_{1}/Fe_{2}O_{3} Single-Atom Catalyst},
author = {Ali Rafsanjani-Abbasi and Florian Buchner and Faith J. Lewis and Lena Puntscher and Florian Kraushofer and Panukorn Sombut and Moritz Eder and Jiří Pavelec and Erik Rheinfrank and Giada Franceschi and Viktor Birschitzky and Michele Riva and Cesare Franchini and Michael Schmid and Ulrike Diebold and Matthias Meier and Georg K. H. Madsen and Gareth S. Parkinson},
url = {https://doi.org/10.1021/acsnano.4c08781},
year = {2024},
date = {2024-09-18},
urldate = {2024-09-18},
journal = {ACS Nano},
volume = {18},
issue = {39},
pages = {26920–26927},
abstract = {Determining the local coordination of the active site is a prerequisite for the reliable modeling of single-atom catalysts (SACs). Obtaining such information is difficult on powder-based systems and much emphasis is placed on density functional theory computations based on idealized low-index surfaces of the support. In this work, we investigate how Pt atoms bind to the (11̅02) facet of α-Fe_{2}O_{3}; a common support material in SACs. Using a combination of scanning tunneling microscopy, X-ray photoelectron spectroscopy, and an extensive computational evolutionary search, we find that Pt atoms significantly reconfigure the support lattice to facilitate a pseudolinear coordination to surface oxygen atoms. Despite breaking three surface Fe–O bonds, this geometry is favored by 0.84 eV over the best configuration involving an unperturbed support. We suggest that the linear O–Pt–O configuration is common in reactive Pt-based SAC systems because it balances thermal stability with the ability to adsorb reactants from the gas phase. Moreover, we conclude that extensive structural searches are necessary to determine realistic active site geometries in single-atom catalysis.},
keywords = {P02, P04, P07, P09},
pubstate = {published},
tppubtype = {article}
}

Wanzenböck, Ralf; Heid, Esther; Riva, Michele; Franceschi, Giada; Imre, Alexander M.; Carrete, Jesús; Diebold, Ulrike; Madsen, Georg K. H.
Exploring Inhomogeneous Surfaces: Ti-rich SrTiO3(110) Reconstructions via Active Learning
Journal ArticleOpen AccessIn: Digital Discovery, vol. 3, pp. 2137-2145, 2024.
Abstract | Links | BibTeX | Tags: P02, P09
@article{Wanzenboeck2024b,
title = {Exploring Inhomogeneous Surfaces: Ti-rich SrTiO_{3}(110) Reconstructions via Active Learning},
author = {Ralf Wanzenböck and Esther Heid and Michele Riva and Giada Franceschi and Alexander M. Imre and Jesús Carrete and Ulrike Diebold and Georg K. H. Madsen},
url = {https://doi.org/10.1039/D4DD00231H},
year = {2024},
date = {2024-09-16},
urldate = {2024-09-16},
journal = {Digital Discovery},
volume = {3},
pages = {2137-2145},
abstract = {The investigation of inhomogeneous surfaces, where various local structures co-exist, is crucial for understanding interfaces of technological interest, yet it presents significant challenges. Here, we study the atomic configurations of the (2×m) Ti-rich surfaces at (110)-oriented SrTiO_{3} by bringing together scanning tunneling microscopy and transferable neural-network force fields combined with evolutionary exploration. We leverage an active learning methodology to iteratively extend the training data as needed for different configurations. Training on only small well-known reconstructions, we are able to extrapolate to the complicated and diverse overlayers encountered in different regions of the heterogeneous SrTiO_{3}(110)-(2×m) surface. Our machine-learning-backed approach generates several new candidate structures, in good agreement with experiment and verified using density functional theory. The approach could be extended to other complex metal oxides featuring large coexisting surface reconstructions.},
keywords = {P02, P09},
pubstate = {published},
tppubtype = {article}
}

Hütner, Johanna I.; Conti, Andrea; Kugler, David; Mittendorfer, Florian; Kresse, Georg; Schmid, Michael; Diebold, Ulrike; Balajka, Jan
Stoichiometric reconstruction of the Al2O3(0001) surface
Journal ArticleOpen AccessarXivIn: Science, vol. 385, pp. 1241–1244, 2024, ISSN: 1095-9203.
Abstract | Links | BibTeX | Tags: P02, P03
@article{Huetner2024,
title = {Stoichiometric reconstruction of the Al_{2}O_{3}(0001) surface},
author = {Johanna I. Hütner and Andrea Conti and David Kugler and Florian Mittendorfer and Georg Kresse and Michael Schmid and Ulrike Diebold and Jan Balajka},
url = {https://www.science.org/doi/10.1126/science.adq4744
https://arxiv.org/abs/2405.19263},
issn = {1095-9203},
year = {2024},
date = {2024-09-12},
urldate = {2024-09-12},
journal = {Science},
volume = {385},
pages = {1241--1244},
publisher = {American Association for the Advancement of Science (AAAS)},
abstract = {Macroscopic properties of materials stem from fundamental atomic-scale details, yet for insulators, resolving surface structures remains a challenge. We imaged the basal (0001) plane of α–aluminum oxide (α-Al_{2}O_{3}) using noncontact atomic force microscopy with an atomically defined tip apex. The surface formed a complex (√31 × √31)R±9° reconstruction. The lateral positions of the individual oxygen and aluminum surface atoms come directly from experiment; we determined with computational modeling how these connect to the underlying crystal bulk. Before the restructuring, the surface Al atoms assume an unfavorable, threefold planar coordination; the reconstruction allows a rehybridization with subsurface O that leads to a substantial energy gain. The reconstructed surface remains stoichiometric, Al_{2}O_{3}.},
keywords = {P02, P03},
pubstate = {published},
tppubtype = {article}
}
Wang, Chunlei; Sombut, Panukorn; Puntscher, Lena; Ulreich, Manuel; Pavelec, Jiri; Rath, David; Balajka, Jan; Meier, Matthias; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare; Parkinson, Gareth S.
A Multitechnique Study of C2H4 Adsorption on a Model Single-Atom Rh1 Catalyst
Journal ArticleOpen AccessIn: The Journal of Physical Chemistry C, vol. 128, iss. 37, pp. 15404–15411, 2024.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Wang_2024b,
title = {A Multitechnique Study of C_{2}H_{4} Adsorption on a Model Single-Atom Rh_{1} Catalyst},
author = {Chunlei Wang and Panukorn Sombut and Lena Puntscher and Manuel Ulreich and Jiri Pavelec and David Rath and Jan Balajka and Matthias Meier and Michael Schmid and Ulrike Diebold and Cesare Franchini and Gareth S. Parkinson},
url = {https://doi.org/10.1021/acs.jpcc.4c03588},
year = {2024},
date = {2024-09-05},
journal = {The Journal of Physical Chemistry C},
volume = {128},
issue = {37},
pages = {15404–15411},
abstract = {Single-atom catalysts are potentially ideal model systems to investigate structure–function relationships in catalysis if the active sites can be uniquely determined. In this work, we study the interaction of C_{2}H_{4} with a model Rh/Fe_{3}O_{4}(001) catalyst that features 2-, 5-, and 6-fold coordinated Rh adatoms, as well as Rh clusters. Using multiple surface-sensitive techniques in combination with calculations of density functional theory (DFT), we follow the thermal evolution of the system and disentangle the behavior of the different species. C_{2}H_{4} adsorption is strongest at the 2-fold coordinated Rh_{1} with a DFT-determined adsorption energy of −2.26 eV. However, desorption occurs at lower temperatures than expected because the Rh migrates into substitutional sites within the support, where the molecule is more weakly bound. The adsorption energy at the 5-fold coordinated Rh sites is predicated to be −1.49 eV, but the superposition of this signal with that from small Rh clusters and additional heterogeneity leads to a broad C_{2}H_{4} desorption shoulder in TPD above room temperature.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Sokolović, Igor; Schmid, Michael; Diebold, Ulrike; Setvín, Martin
How to cleave cubic perovskite oxides
Journal ArticleOpen AccessSubmittedarXivIn: arXiv, 2024.
Abstract | Links | BibTeX | Tags: P02
@article{Sokolovic_2024a,
title = {How to cleave cubic perovskite oxides},
author = {Igor Sokolović and Michael Schmid and Ulrike Diebold and Martin Setvín},
url = {https://arxiv.org/abs/2408.08996},
year = {2024},
date = {2024-08-16},
journal = {arXiv},
abstract = {Surfaces of cubic perovskite oxides attract significant attention for their physical tunability and high potential for technical applications. Bulk-terminated surfaces are desirable for theoretical modelling and experimental reproducibility, yet there is a lack of methods for preparing such well-defined surfaces. We discuss a method for strain-assisted cleaving of perovskite single crystals, using a setup easily transferable between different experimental systems. The details of the cleaving device and the procedure were optimized in a systematic study on the model SrTiO_{3}. The large-area morphology and typical distribution of surface terminations on cleaved SrTiO_{3}(001) is presented, with specific guidelines on how to distinguish well-cleaved surfaces from conchoidally fractured ones. The cleaving is applicable to other cubic perovskites, as demonstrated on KTaO_{3}(001) and BaTiO_{3}(001). This approach opens up a pathway for obtaining high-quality surfaces of this promising class of materials.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
Kraushofer, Florian; Imre, Alexander M.; Franceschi, Giada; Kißlinger, Tilman; Rheinfrank, Erik; Schmid, Michael; Diebold, Ulrike; Hammer, Lutz; Riva, Michele
ViPErLEED package I: Calculation of I(V) curves and structural optimization
Journal ArticleOpen AccessAccepted ArticlearXivIn: Physical Review Research, 2024.
Abstract | Links | BibTeX | Tags: P02
@article{Kraushofer_2024a,
title = {ViPErLEED package I: Calculation of I(V) curves and structural optimization},
author = {Florian Kraushofer and Alexander M. Imre and Giada Franceschi and Tilman Kißlinger and Erik Rheinfrank and Michael Schmid and Ulrike Diebold and Lutz Hammer and Michele Riva},
url = {https://arxiv.org/abs/2406.18821},
year = {2024},
date = {2024-06-27},
urldate = {2024-06-27},
journal = {Physical Review Research},
abstract = {Low-energy electron diffraction (LEED) is a widely used technique in surface-science. Yet, it is rarely used to its full potential. The quantitative information about the surface structure, contained in the modulation of the intensities of the diffracted beams as a function of incident electron energy, LEED I(V), is underutilized. To acquire these data, minor adjustments would be required in most experimental setups, but existing analysis software is cumbersome to use. ViPErLEED (Vienna package for Erlangen LEED) lowers these barriers, introducing a combined solution for data acquisition, extraction, and computational analysis. These parts are discussed in three separate publications. Here, the focus is on the computational part of ViPErLEED, which performs automated LEED-I(V) calculations and structural optimization. Minimal user input is required, and the functionality is significantly enhanced compared to existing solutions. Computation is performed by embedding the Erlangen tensor-LEED package (TensErLEED). ViPErLEED manages parallelization, monitors convergence, and processes input and output. This makes LEED I(V) more accessible to new users while minimizing the potential for errors and the manual labor. Added functionality includes structure-dependent defaults, automatic detection of bulk and surface symmetries and their relationship, automated symmetry-preserving search procedures, adjustments to the TensErLEED code to handle larger systems, as well as parallelization and optimization. Modern file formats are used as input and output, and there is a direct interface to the Atomic Simulation Environment (ASE) package. The software is implemented primarily in Python (version >=3.7) and provided as an open-source package (GNU GPLv3 or later). A structure determination of the α-Fe_{2}O_{3}(1-102)-(1x1) surface is presented as an example for the application of the software.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
Schmid, Michael; Kraushofer, Florian; Imre, Alexander M.; Kißlinger, Tilman; Hammer, Lutz; Diebold, Ulrike; Riva, Michele
ViPErLEED package II: Spot tracking, extraction and processing of I(V) curves
Journal ArticleOpen AccessAccepted ArticlearXivIn: Physical Review Research, 2024.
Abstract | Links | BibTeX | Tags: P02
@article{Schmid_2024a,
title = {ViPErLEED package II: Spot tracking, extraction and processing of I(V) curves},
author = {Michael Schmid and Florian Kraushofer and Alexander M. Imre and Tilman Kißlinger and Lutz Hammer and Ulrike Diebold and Michele Riva},
url = {https://arxiv.org/abs/2406.18413},
year = {2024},
date = {2024-06-26},
urldate = {2024-06-26},
journal = {Physical Review Research},
abstract = {As part of the ViPErLEED project (Vienna package for Erlangen LEED, low-energy electron diffraction), computer programs have been developed for facile and user-friendly data extraction from movies of LEED images. The programs make use of some concepts from astronomical image processing and analysis. As a first step, flat-field and dark-frame corrections reduce the effects of inhomogeneities of the camera and screen. In a second step, for identifying all diffraction maxima ("spots"), it is sufficient to manually mark and label a single spot or very few spots. Then the program can automatically identify all other spots and determine the distortions of the image. This forms the basis for automatic spot tracking (following the "beams" as they move across the LEED screen) and intensity measurement. Even for complex structures with hundreds to a few thousand diffraction beams, this step takes less than a minute. The package also includes a program for further processing of these I(V) curves (averaging of equivalent beams, manual and/or automatic selection, smoothing) as well as several utilities. The software is implemented as a set of plugins for the public-domain image processing program ImageJ and provided as an open-source package.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
Rath, David; Mikerásek, Vojtěch; Wang, Chunlei; Eder, Moritz; Schmid, Michael; Diebold, Ulrike; Parkinson, Gareth S.; Pavelec, Jiří
Journal ArticleOpen AccessIn: Review of Scientific Instruments, vol. 95, iss. 6, pp. 065106, 2024.
Abstract | Links | BibTeX | Tags: P02, P04
@article{Rath2024,
title = {Infrared Reflection Absorption Spectroscopy Setup with Incidence Angle Selection for Surfaces of Non-Metals},
author = {David Rath and Vojtěch Mikerásek and Chunlei Wang and Moritz Eder and Michael Schmid and Ulrike Diebold and Gareth S. Parkinson and Jiří Pavelec},
url = {https://arxiv.org/abs/2403.19263},
doi = {https://doi.org/10.1063/5.0210860},
year = {2024},
date = {2024-06-07},
urldate = {2024-06-07},
journal = {Review of Scientific Instruments},
volume = {95},
issue = {6},
pages = {065106},
publisher = {arXiv},
abstract = {Infrared Reflection Absorption Spectroscopy (IRAS) on dielectric single crystals is challenging because the optimal incidence angles for light-adsorbate interaction coincide with regions of low IR reflectivity. Here, we introduce an optimized IRAS setup that maximizes the signal-to-noise ratio for non-metals. This is achieved by maximizing light throughput, and by selecting optimal incidence angles that directly impact the peak heights in the spectra. The setup uses a commercial FTIR spectrometer and is usable in ultra-high vacuum (UHV). Specifically, the design features sample illumination and collection mirrors with a high numerical aperture inside the UHV system, and an adjustable aperture to select the incidence angle range on the sample. This is important for p-polarized measurements on dielectrics, because the peaks in the spectra reverse direction at the Brewster angle (band inversion). The system components are connected precisely via a single flange, ensuring long-term stability. We studied the signal-to-noise (SNR) variation in p-polarized IRAS spectra for one monolayer of CO on TiO_{2}(110) as a function of incidence angle range, where a maximum signal-to-noise ratio of 70 was achieved at 4 cm^{-1} resolution in five minutes measurement time. The capabilities for s-polarization are demonstrated by measuring one monolayer D_{2}O adsorbed on a TiO_{2}(110) surface, where a SNR of 65 was achieved at a delta_R/R0 peak height of 1.4x10-4 in twenty minutes.},
keywords = {P02, P04},
pubstate = {published},
tppubtype = {article}
}