Taming complexity in polaron configurational space: TACO scientists present a new machine-learning approach to accelerate polaron distribution calculation in recently published npj Computational Materials publication.
Polarons are quasiparticles that arise from the interaction of charges (electrons or holes) with the lattice vibrations (phonons) in a crystal. Polarons are a pervasive phenomenon in transition-metal oxides. They have a strong impact on the properties and functionality of materials that are important in many technical applications. For many years, polarons have been at the focus of theoretical research to understand better and predict those material properties. Despite the huge importance and considerable efforts, detailed predictions have been stalled simply by the overwhelmingly large number of possible configurations that polarons can occupy in a crystal lattice.
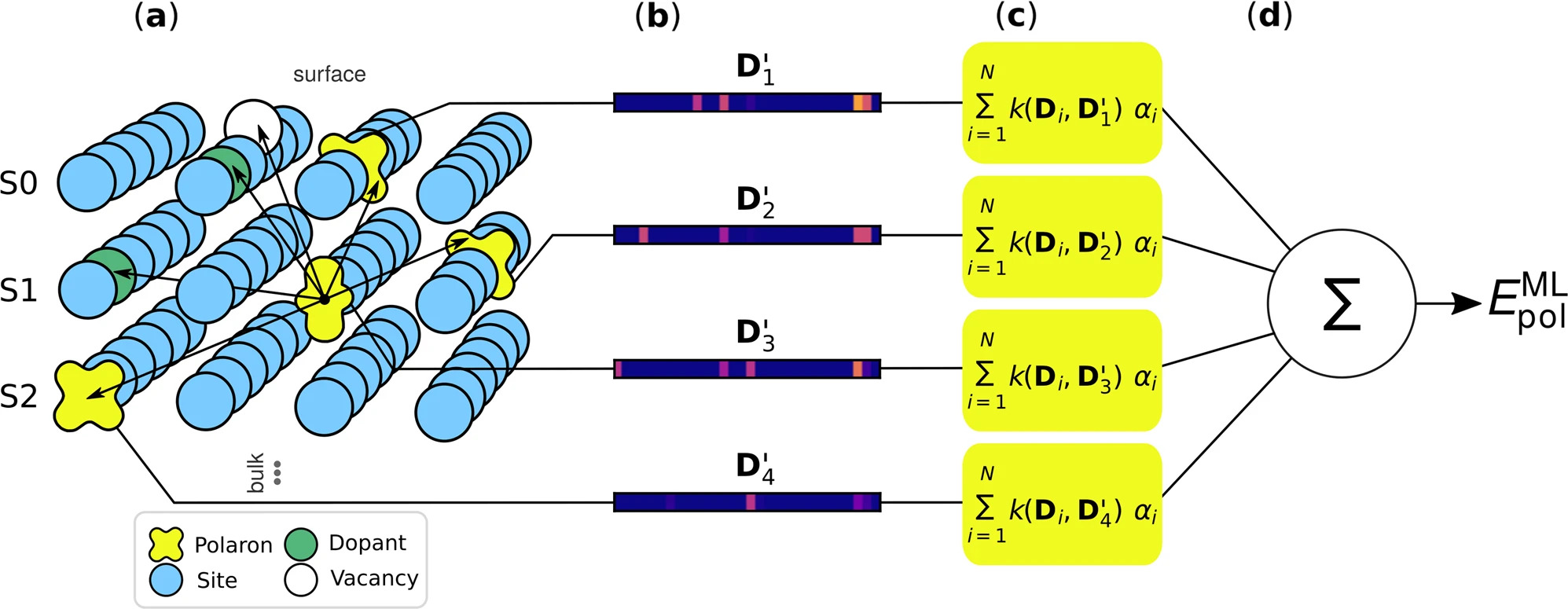
A team of TACO scientists has now accepted the challenge to tame this complexity: In a just-published paper, the team led by Ph.D. students Viktor Birschitzky and Florian Ellinger, under the guidance of the TACO Co-PI Michele Reticcioli and PIs Cesare Franchini and Ulrike Diebold, present a new machine-learning (ML) approach to accelerating significantly the previously prohibitively demanding calculations. The scientists trained the ML algorithm on a small sample of detailed yet computationally expensive DFT first-principles calculations (molecular dynamics and random sampling) of polaron configurations. Later, the algorithm explored the vast configurational space to identify energetically favourable and thus more likely polaron arrangements at variable polaron concentrations. Where less than 500 polaron configurations could be calculated with previous first-principle approaches, millions of structures can now be scanned and explored with the new ML approach.
The researchers validated the approach on two well-known material surface systems: rutile TiO2 (110), and Nb-doped perovskite SrTiO3 (001). Particularly for this latter surface system, the ML-aided procedure achieved a systematic improvement of the polaronic ground state configurations compared to the more traditional approaches. The researchers are optimistic that the algorithm can be adapted relatively easily to consider other types of crystal defects, such as adatoms. Also, the scalability of the code for direct comparison of the simulation results with experimental data is supposed to be good. This would open the path for applying the code to many more material systems.
The paper was published open-access on June 6 in npj Computational Materials, a member of the Nature family of journals.