
Surface structure and reactivity of
multi-component oxides at the atomic scale
Subproject P02
Multi-component metal oxides exhibit a plethora of stoichiometry-dependent structural phases at the surface, even if the composition of the bulk is kept the same. The long-term objective of P02 is to unravel the relationship between surface electronic and geometric structure and reactivity, to ultimately tune these materials for energy-related reactions such as the ORR. The project applies the surface science approach. We will grow well-defined, epitaxial perovskite thin films of LSFO and LSMO in a UHV-based PLD/surface science apparatus under tight control of the surface stoichiometry in the first project period. We will determine the coordinates of surface atoms quantitatively using LEED-IV in close collaboration with theoretical groups.
Theoretical models will also help with interpreting atomically-resolved ncAFM/STM images. These images give direct insights into the behavior of polarons in these complex materials and show how adsorbates such as O2, H2O, CO, and CO2 interact with electronic and structural defects. XPS, TPD, and FTIR of these well-defined systems will deliver desorption energies, vibrational frequencies, and spectral fingerprints. These experimental data on well-defined systems will build a bridge when tested under ‘realistic’ environments at high pressure/temperature and in aqueous solutions. They will also serve to validate ML-based theory approaches.


Ulrike Diebold
PI
Expertise
Our expertise is experimental surface science. We operate a total of seven ultrahigh-vacuum (UHV) chambers, which contain virtually all main experimental surface science techniques, as well as an (electro-)chemistry lab.
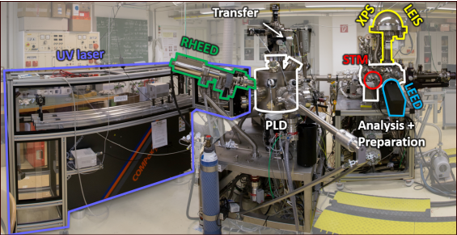
All chambers are equipped with facilities for sample preparation (sputtering/annealing/gas dosing), as well as various growth techniques (e-beam evaporators, Knudsen cells, UHV-compatible sputter deposition, pulsed laser deposition (PLD)).
Analysis techniques used in our research include:
- Scanning Tunneling Microscopy (STM) (in UHV 4K – 300 K, electrochemical STM)
- Atomic Force Microscopy (AFM): UHV-based (q+ sensor) and in the ambient (cantilever-based)
- Low-Energy Electron Diffraction (LEED)
- Reflection High Energy Diffraction (RHEED)
- X-ray Photoelectron Spectroscopy (XPS)
- Ultraviolet Photoelectron Spectroscopy (UPS)
- Auger Electron Spectroscopy (AES)
- Low-energy He+ ion scattering (LEIS)
- Thermal Programmed Desorption Spectroscopy (TPD)
Of particular use for TACO is our combined Pulsed-Laser Deposition/Surface Science setup shown on the right-hand side. It allows the growth of multi-component metal oxides and the investigations of surface properties within the same UHV setup.
Team

Ulrike Diebold
PI

Michele Riva
co-PI

Michael Schmid
co-PI

Florian Dörr
PhD Student

Alexander Imre
PhD Student, Student Representative 23–24

Paul Haidegger
Master Student

Marie Kienzer
Master Student
Associates

Sarah Tobisch
PhD Student
Former Members

Michael Brunthaler
Master Student

Erik Rheinfrank
PhD-Student

Christoph Schattauer
PostDoc
Publications
2022
Schmid, Michael; Parkinson, Gareth S.; Diebold, Ulrike
Analysis of Temperature-Programmed Desorption via Equilibrium Thermodynamics
Journal ArticleOpen AccessIn: ACS Physical Chemistry Au, vol. 3, iss. 1, pp. 44–62, 2022.
Abstract | Links | BibTeX | Tags: P02, P04
@article{Schmid2022,
title = {Analysis of Temperature-Programmed Desorption via Equilibrium Thermodynamics},
author = {Michael Schmid and Gareth S. Parkinson and Ulrike Diebold},
doi = {10.1021/acsphyschemau.2c00031},
year = {2022},
date = {2022-11-15},
journal = {ACS Physical Chemistry Au},
volume = {3},
issue = {1},
pages = {44--62},
publisher = {American Chemical Society (ACS)},
abstract = {Temperature-programmed desorption (TPD) experiments in surface science are usually analyzed using the Polanyi–Wigner equation and/or transition-state theory. These methods are far from straightforward, and the determination of the pre-exponential factor is often problematic. We present a different method based on equilibrium thermodynamics, which builds on an approach previously used for TPD by Kreuzer et al. (\textit{Surf. Sci.}\textbf{1988}). Equations for the desorption rate are presented for three different types of surface–adsorbate interactions: (i) a 2D ideal hard-sphere gas with a negligible diffusion barrier, (ii) an ideal lattice gas, that is, fixed adsorption sites without interaction between the adsorbates, and (iii) a lattice gas with a distribution of (site-dependent) adsorption energies. We show that the coverage dependence of the sticking coefficient for adsorption at the desorption temperature determines whether the desorption process can be described by first- or second-order kinetics. The sticking coefficient at the desorption temperature must also be known for a quantitative determination of the adsorption energy, but it has a rather weak influence (like the pre-exponential factor in a traditional TPD analysis). Quantitative analysis is also influenced by the vibrational contributions to the energy and entropy. For the case of a single adsorption energy, we provide equations to directly convert peak temperatures into adsorption energies. These equations also provide an approximation of the desorption energy in cases that cannot be described by a fixed pre-exponential factor. For the case of a distribution of adsorption energies, the desorption spectra cannot be considered a superposition of desorption spectra corresponding to the different energies. Nevertheless, we present a method to extract the distribution of adsorption energies from TPD spectra, and we rationalize the energy resolution of TPD experiments. The analytical results are complemented by a program for simulation and analysis of TPD data.},
keywords = {P02, P04},
pubstate = {published},
tppubtype = {article}
}
Wang, Zhichang; Reticcioli, Michele; Jakub, Zdenek; Sokolović, Igor; Meier, Matthias; Boatner, Lynn A; Schmid, Michael; Parkinson, Gareth S.; Diebold, Ulrike; Franchini, Cesare; Setvin, Martin
Surface chemistry on a polarizable surface: Coupling of CO with KTaO 3(001)
Journal ArticleOpen AccessIn: Science Advances, vol. 8, iss. 33, 2022.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Wang2022,
title = {Surface chemistry on a polarizable surface: Coupling of CO with KTaO _{3}(001)},
author = {Zhichang Wang and Michele Reticcioli and Zdenek Jakub and Igor Sokolović and Matthias Meier and Lynn A Boatner and Michael Schmid and Gareth S. Parkinson and Ulrike Diebold and Cesare Franchini and Martin Setvin},
url = {https://www.science.org/doi/10.1126/sciadv.abq1433},
doi = {10.1126/sciadv.abq1433},
year = {2022},
date = {2022-08-19},
urldate = {2022-08-19},
journal = {Science Advances},
volume = {8},
issue = {33},
publisher = {American Association for the Advancement of Science (AAAS)},
abstract = {Polarizable materials attract attention in catalysis because they have a free parameter for tuning chemical reactivity. Their surfaces entangle the dielectric polarization with surface polarity, excess charge, and orbital hybridization. How this affects individual adsorbed molecules is shown for the incipient ferroelectric perovskite KTaO_{3}. This intrinsically polar material cleaves along (001) into KO- and TaO_{2}-terminated surface domains. At TaO_{2} terraces, the polarity-compensating excess electrons form a two-dimensional electron gas and can also localize by coupling to ferroelectric distortions. TaO_{2} terraces host two distinct types of CO molecules, adsorbed at equivalent lattice sites but charged differently as seen in atomic force microscopy/scanning tunneling microscopy. Temperature-programmed desorption shows substantially stronger binding of the charged CO; in density functional theory calculations, the excess charge favors a bipolaronic configuration coupled to the CO. These results pinpoint how adsorption states couple to ferroelectric polarization.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Reticcioli, Michele; Wang, Zhichang; Schmid, Michael; Wrana, Dominik; Boatner, Lynn A.; Diebold, Ulrike; Setvin, Martin; Franchini, Cesare
Competing electronic states emerging on polar surfaces
Journal ArticleOpen AccessIn: Nature Communications, vol. 13, no. 4311, 2022.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Reticcioli2022,
title = {Competing electronic states emerging on polar surfaces},
author = {Michele Reticcioli and Zhichang Wang and Michael Schmid and Dominik Wrana and Lynn A. Boatner and Ulrike Diebold and Martin Setvin and Cesare Franchini},
url = {https://www.nature.com/articles/s41467-022-31953-6},
doi = {10.1038/s41467-022-31953-6},
year = {2022},
date = {2022-07-25},
urldate = {2022-07-25},
journal = {Nature Communications},
volume = {13},
number = {4311},
publisher = {Springer Science and Business Media LLC},
abstract = {Excess charge on polar surfaces of ionic compounds is commonly described by the two-dimensional electron gas (2DEG) model, a homogeneous distribution of charge, spatially-confined in a few atomic layers. Here, by combining scanning probe microscopy with density functional theory calculations, we show that excess charge on the polar TaO_{2} termination of KTaO_{3}(001) forms more complex electronic states with different degrees of spatial and electronic localization: charge density waves (CDW) coexist with strongly-localized electron polarons and bipolarons. These surface electronic reconstructions, originating from the combined action of electron-lattice interaction and electronic correlation, are energetically more favorable than the 2DEG solution. They exhibit distinct spectroscopy signals and impact on the surface properties, as manifested by a local suppression of ferroelectric distortions.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
Birschitzky, Viktor C; Ellinger, Florian; Diebold, Ulrike; Reticcioli, Michele; Franchini, Cesare
Machine learning for exploring small polaron configurational space
Journal ArticleOpen AccessIn: npj Computational Materials, vol. 8, no. 125, 2022.
Abstract | Links | BibTeX | Tags: P02, P07
@article{Birschitzky2022,
title = {Machine learning for exploring small polaron configurational space},
author = {Viktor C Birschitzky and Florian Ellinger and Ulrike Diebold and Michele Reticcioli and Cesare Franchini},
url = {https://www.nature.com/articles/s41524-022-00805-8},
doi = {10.1038/s41524-022-00805-8},
year = {2022},
date = {2022-06-06},
urldate = {2022-06-06},
journal = {npj Computational Materials},
volume = {8},
number = {125},
publisher = {Springer Science and Business Media LLC},
abstract = {Polaron defects are ubiquitous in materials and play an important role in many processes involving carrier mobility, charge transfer and surface reactivity. Determining small polarons’ spatial distributions is essential to understand materials properties and functionalities. However, the required exploration of the configurational space is computationally demanding when using first principles methods. Here, we propose a machine-learning (ML) accelerated search that determines the ground state polaronic configuration. The ML model is trained on databases of polaron configurations generated by density functional theory (DFT) via molecular dynamics or random sampling. To establish a mapping between configurations and their stability, we designed descriptors modelling the interactions among polarons and charged point defects. We used the DFT+ML protocol to explore the polaron configurational space for two surface-systems, reduced rutile TiO_{2}(110) and Nb-doped SrTiO_{3}(001). The ML-aided search proposes additional polaronic configurations and can be utilized to determine optimal polaron distributions at any charge concentration.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
Merte, Lindsay R; Bisbo, Malthe Kjær; Sokolović, Igor; Setvín, Martin; Hagman, Benjamin; Shipilin, Mikhail; Schmid, Michael; Diebold, Ulrike; Lundgren, Edvin; Hammer, Bjørk
Journal ArticleOpen AccessIn: Angewandte Chemie - International Edition, vol. 61, iss. 25, pp. e202204244, 2022.
Abstract | Links | BibTeX | Tags: P02
@article{ANGEWANDTE2022,
title = {Structure of an Ultrathin Oxide on Pt_{3}Sn(111) Solved by Machine Learning Enhanced Global Optimization},
author = {Lindsay R Merte and Malthe Kjær Bisbo and Igor Sokolović and Martin Setvín and Benjamin Hagman and Mikhail Shipilin and Michael Schmid and Ulrike Diebold and Edvin Lundgren and Bjørk Hammer},
url = {https://onlinelibrary.wiley.com/doi/10.1002/anie.202204244},
doi = {10.1002/anie.202204244},
year = {2022},
date = {2022-04-05},
urldate = {2022-04-05},
journal = {Angewandte Chemie - International Edition},
volume = {61},
issue = {25},
pages = {e202204244},
abstract = {Determination of the atomic structure of solid surfaces typically depends on comparison of measured properties with simulations based on hypothesized structural models. For simple structures, the models may be guessed, but for more complex structures there is a need for reliable theory-based search algorithms. So far, such methods have been limited by the combinatorial complexity and computational expense of sufficiently accurate energy estimation for surfaces. However, the introduction of machine learning methods has the potential to change this radically. Here, we demonstrate how an evolutionary algorithm, utilizing machine learning for accelerated energy estimation and diverse population generation, can be used to solve an unknown surface structure—the (4×4) surface oxide on Pt_{3}Sn(111)–based on limited experimental input. The algorithm is efficient and robust, and should be broadly applicable in surface studies, where it can replace manual, intuition based model generation.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
Meier, Matthias; Hulva, Jan; Jakub, Zdenek; Kraushofer, Florian; Bobić, Mislav; Bliem, Roland; Setvin, Martin; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare; Parkinson, Gareth S.
Journal ArticleOpen AccessIn: Science Advances, vol. 8, iss. 13, pp. eabn4580, 2022.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{SCIADV2022,
title = {CO oxidation by Pt_{2}/Fe_{3}O_{4}: Metastable dimer and support configurations facilitate lattice oxygen extraction},
author = {Matthias Meier and Jan Hulva and Zdenek Jakub and Florian Kraushofer and Mislav Bobić and Roland Bliem and Martin Setvin and Michael Schmid and Ulrike Diebold and Cesare Franchini and Gareth S. Parkinson},
url = {https://www.science.org/doi/10.1126/sciadv.abn4580},
doi = {10.1126/sciadv.abn4580},
year = {2022},
date = {2022-04-01},
urldate = {2022-04-01},
journal = {Science Advances},
volume = {8},
issue = {13},
pages = {eabn4580},
abstract = {Heterogeneous catalysts based on subnanometer metal clusters often exhibit strongly size-dependent properties, and the addition or removal of a single atom can make all the difference. Identifying the most active species and deciphering the reaction mechanism is extremely difficult, however, because it is often not clear how the catalyst evolves in operando. Here, we use a combination of atomically resolved scanning probe microscopies, spectroscopic techniques, and density functional theory (DFT)–based calculations to study CO oxidation by a model Pt/Fe_{3}O_{4}(001) “single-atom” catalyst. We demonstrate that (PtCO)_{2} dimers, formed dynamically through the agglomeration of mobile Pt-carbonyl species, catalyze a reaction involving the oxide support to form CO_{2}. Pt_{2} dimers produce one CO_{2} molecule before falling apart into two adatoms, releasing the second CO. Olattice extraction only becomes facile when both the Pt-dimer and the Fe_{3}O_{4} support can access metastable configurations, suggesting that substantial, concerted rearrangements of both cluster and support must be considered for reactions occurring at elevated temperature.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}
Reticcioli, Michele; Diebold, Ulrike; Franchini, Cesare
Modeling polarons in density functional theory: lessons learned from TiO2
Journal ArticleOpen AccessIn: Journal of Physics: Condensed Matter, vol. 34, no. 20, pp. 204006, 2022.
Abstract | Links | BibTeX | Tags: P02, P07
@article{JPCM2022,
title = {Modeling polarons in density functional theory: lessons learned from TiO_{2}},
author = {Michele Reticcioli and Ulrike Diebold and Cesare Franchini},
url = {https://iopscience.iop.org/article/10.1088/1361-648X/ac58d7},
doi = {10.1088/1361-648X/ac58d7},
year = {2022},
date = {2022-03-14},
urldate = {2022-03-14},
journal = {Journal of Physics: Condensed Matter},
volume = {34},
number = {20},
pages = {204006},
abstract = {Density functional theory (DFT) is nowadays one of the most broadly used and successful techniques to study the properties of polarons and their effects in materials. Here, we systematically analyze the aspects of the theoretical calculations that are crucial to obtain reliable predictions in agreement with the experimental observations. We focus on rutile TiO_{2}, a prototypical polaronic compound, and compare the formation of polarons on the (110) surface and subsurface atomic layers. As expected, the parameter U used to correct the electronic correlation in the DFT+U formalism affects the resulting charge localization, local structural distortions and electronic properties of polarons. Moreover, the polaron localization can be driven to different sites by strain: Due to different local environments, surface and subsurface polarons show different responses to the applied strain, with impact on the relative energy stability. An accurate description of the properties of polarons is key to understand their impact on complex phenomena and applications: As an example, we show the effects of lattice strain on the interaction between polarons and CO adsorbates.},
keywords = {P02, P07},
pubstate = {published},
tppubtype = {article}
}
Franceschi, Giada; Schmid, Michael; Diebold, Ulrike; Riva, Michele
Reconstruction changes drive surface diffusion and determine the flatness of oxide surfaces
Journal ArticleOpen AccessIn: Journal of Vacuum Science & Technology A, vol. 40, no. 2, pp. 023206, 2022.
Abstract | Links | BibTeX | Tags: P02
@article{Franceschi2022,
title = {Reconstruction changes drive surface diffusion and determine the flatness of oxide surfaces},
author = {Giada Franceschi and Michael Schmid and Ulrike Diebold and Michele Riva},
doi = {10.1116/6.0001704},
year = {2022},
date = {2022-02-22},
urldate = {2022-02-22},
journal = {Journal of Vacuum Science & Technology A},
volume = {40},
number = {2},
pages = {023206},
publisher = {American Vacuum Society},
abstract = {Surface diffusion on metal oxides is key in many areas of materials technology, yet it has been scarcely explored at the atomic scale. This work provides phenomenological insights from scanning tunneling microscopy on the link between surface diffusion, surface atomic structure, and oxygen chemical potential based on three model oxide surfaces: Fe}2}O_{3}(1-102), La_{1−x}Sr_{x}MnO_{3}(110), and In_{2}O_{3}(111). In all instances, changing the oxygen chemical potential used for annealing stabilizes reconstructions of different compositions while promoting the flattening of the surface morphology—a sign of enhanced surface diffusion. It is argued that thermodynamics, rather than kinetics, rules surface diffusion under these conditions: the composition change of the surface reconstructions formed at differently oxidizing conditions drives mass transport across the surface.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
Schmid, Michael; Rath, David; Diebold, Ulrike
Why and How Savitzky–Golay Filters Should Be Replaced
Journal ArticleOpen AccessIn: ACS Measurement Science Au, vol. 2, no. 2, pp. 185–196, 2022.
Abstract | Links | BibTeX | Tags: P02
@article{ACSMEASURE2022,
title = {Why and How Savitzky–Golay Filters Should Be Replaced},
author = {Michael Schmid and David Rath and Ulrike Diebold},
url = {https://pubs.acs.org/doi/10.1021/acsmeasuresciau.1c00054},
doi = {10.1021/acsmeasuresciau.1c00054},
year = {2022},
date = {2022-02-17},
urldate = {2022-02-17},
journal = {ACS Measurement Science Au},
volume = {2},
number = {2},
pages = {185--196},
abstract = {Savitzky–Golay (SG) filtering, based on local least-squares fitting of the data by polynomials, is a popular method for smoothing data and calculations of derivatives of noisy data. At frequencies above the cutoff, SG filters have poor noise suppression; this unnecessarily reduces the signal-to-noise ratio, especially when calculating derivatives of the data. In addition, SG filtering near the boundaries of the data range is prone to artifacts, which are especially strong when using SG filters for calculating derivatives of the data. We show how these disadvantages can be avoided while keeping the advantageous properties of SG filters. We present two classes of finite impulse response (FIR) filters with substantially improved frequency response: (i) SG filters with fitting weights in the shape of a window function and (ii) convolution kernels based on the sinc function with a Gaussian-like window function and additional corrections for improving the frequency response in the passband (modified sinc kernel). Compared with standard SG filters, the only price to pay for the improvement is a moderate increase in the kernel size. Smoothing at the boundaries of the data can be improved with a non-FIR method, the Whittaker–Henderson smoother, or by linear extrapolation of the data, followed by convolution with a modified sinc kernel, and we show that the latter is preferable in most cases. We provide computer programs and equations for the smoothing parameters of these smoothers when used as plug-in replacements for SG filters and describe how to choose smoothing parameters to preserve peak heights in spectra.},
keywords = {P02},
pubstate = {published},
tppubtype = {article}
}
2021
Jakub, Zdenek; Meier, Matthias; Kraushofer, Florian; Balajka, Jan; Pavelec, Jiri; Schmid, Michael; Franchini, Cesare; Diebold, Ulrike; Parkinson, Gareth S.
Rapid oxygen exchange between hematite and water vapor
Journal ArticleOpen AccessIn: Nature Communications, vol. 12, iss. 1, no. 6488, 2021.
Abstract | Links | BibTeX | Tags: P02, P04, P07
@article{Jakub2021,
title = {Rapid oxygen exchange between hematite and water vapor},
author = {Zdenek Jakub and Matthias Meier and Florian Kraushofer and Jan Balajka and Jiri Pavelec and Michael Schmid and Cesare Franchini and Ulrike Diebold and Gareth S. Parkinson},
doi = {10.1038/s41467-021-26601-4},
year = {2021},
date = {2021-11-10},
journal = {Nature Communications},
volume = {12},
number = {6488},
issue = {1},
publisher = {Springer Science and Business Media LLC},
abstract = {Oxygen exchange at oxide/liquid and oxide/gas interfaces is important in technology and environmental studies, as it is closely linked to both catalytic activity and material degradation. The atomic-scale details are mostly unknown, however, and are often ascribed to poorly defined defects in the crystal lattice. Here we show that even thermodynamically stable, well-ordered surfaces can be surprisingly reactive. Specifically, we show that all the 3-fold coordinated lattice oxygen atoms on a defect-free single-crystalline “r-cut” (1-102) surface of hematite (α-Fe_{2}O_{3}) are exchanged with oxygen from surrounding water vapor within minutes at temperatures below 70 °C, while the atomic-scale surface structure is unperturbed by the process. A similar behavior is observed after liquid-water exposure, but the experimental data clearly show most of the exchange happens during desorption of the final monolayer, not during immersion. Density functional theory computations show that the exchange can happen during on-surface diffusion, where the cost of the lattice oxygen extraction is compensated by the stability of an HO-HOH-OH complex. Such insights into lattice oxygen stability are highly relevant for many research fields ranging from catalysis and hydrogen production to geochemistry and paleoclimatology.},
keywords = {P02, P04, P07},
pubstate = {published},
tppubtype = {article}
}