
Catalysis by ultrathin
LaBO3 (B=Co, Fe) perovskite films
Subproject P08
Perovskites are important catalysts, but detailed knowledge of their surface structure and chemistry is often lacking. The long-term objective of P08 is to elucidate structure-function correlations and visualize molecule-perovskite interaction in reactions involving O2, H2, CO, CO2, or H2O.
In the first project period, we will develop surface science-based model systems of LaCoO3 and LaFeO3 perovskites. Both epitaxial and polycrystalline thin films will be grown in UHV, guided by characterization via LEED, SXRD, SEM/EBSD, XPS/UPS/LEIS, IRAS, and TPD. Isotopically (18O or 13C) labeled adsorbates or films will reveal how oxygen and oxygen-containing molecules are activated. We will analyze the data in close collaboration with theoretical groups who simulate structure, stability, and infrared spectra (P03 Kresse).
We will employ a unique combination of in situ surface spectroscopy (PM-IRAS, NAP-XPS, SXRD) and in situ surface microscopy (PEEM, SPEM), combined with MS gas phase analysis, to monitor ongoing reactions from HV to atmospheric pressure. This procedure should enable us to gain fundamental insights into the interplay of ternary oxide atomic and electronic structure, defects, composition, adsorption, as well as initiation and spatial progression of surface reactions on the mesoscale via reaction fronts (local kinetics by imaging). Project P08 will create the required bridge between single crystals (P02 Diebold, P04 Parkinson) and more application-relevant nanomaterials (P10 Föttinger).

Günther Rupprechter
PI
Expertise
Our expertise is experimental surface science and its application to studies in heterogeneous catalysis. We operate a total of seven ultrahigh-vacuum (UHV) chambers, three of which are coupled to high-pressure cells. In situ and operando studies of surface reactions are carried out by area-averaging surface spectroscopy and real-time surface microscopy on the nano- and mesoscale. All chambers are equipped with facilities for sample preparation (sputtering, annealing, gas dosing), as well as various growth techniques (e-beam evaporators, Knudsen cells, sputter deposition). Analysis techniques used in our research include:
- Auger Electron Spectroscopy (AES)
- Field Emission Microscopy (FEM)
- Field Ion Microscopy (FIM)
- Gas Chromatography (GC)
- Low-Energy Electron Diffraction (LEED)
- Low-Energy Ion Scattering (LEIS)
- Mass Spectroscopy (MS)
- PhotoEmission Electron Microscopy (PEEM)
- Polarization Modulation Infrared Reflection-Absorption Spectroscopy (PM-IRAS)
- Sum Frequency Generation (SFG)
- Scanning PhotoElectron Microscopy (SPEM)
- Scanning Tunneling Microscopy (STM)
- Temperature-Programmed Desorption (TPD)
- Ultraviolet Photoelectron Spectroscopy (UPS)
- X-ray Absorption Spectroscopy (XAS)
- Surface X-Ray Diffraction (SXRD)
- X-ray Photoelectron Spectroscopy (XPS)
Collaboration Partners:
- Prof. Andreas Stierle, DESY Hamburg, Germany: SXRD
- Dr. Luca Gregoratti, ELETTRA Sincrotrone Trieste, Italy: SPEM
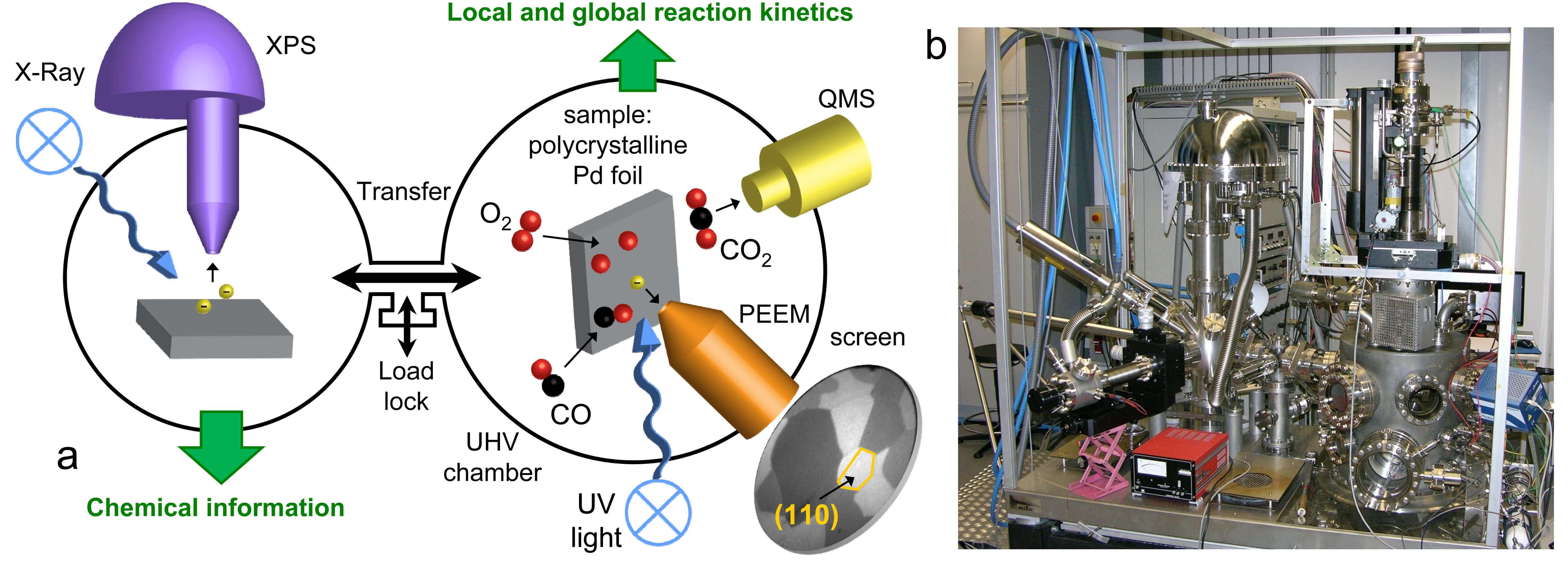
Schematic overview on the left (a); photograph on the right (b)
The combined application of photoemission electron microscopy (PEEM) and scanning photoelectron microscopy (SPEM) is particularly beneficial for TACO because these techniques allow visualizing ongoing reactions and local surface analysis on a µm-scale.
Team

Günther Rupprechter
PI

Qaisar Maqbool
PostDoc

Thomas Wicht
PhD Student

Parinya Tangpakonsab PhD Student
Associates

Alexander Genest
PostDoc

Thomas Haunold
PostDoc

Xia Li
PostDoc

Philipp Winkler
PostDoc

Nevzat Yigit
PostDoc

Johannes Zeininger
PostDoc

Maximilian Raab
PhD Student
Former Members

Yvan J. O. Asencios
Visiting Professor

Camilla Ferreira de Sá Codeço
Visiting Professor

Yuri Suchorski
Associated Professor
Publications
2019
Singraber, Andreas; Morawietz, Tobias; Behler, Jörg; Dellago, Christoph
Parallel Multistream Training of High-Dimensional Neural Network Potentials
Journal ArticleIn: Journal of Chemical Theory and Computation, vol. 15, no. 5, pp. 3075–3092, 2019.
Abstract | Links | BibTeX | Tags: P12, pre-TACO
@article{Singraber2019,
title = {Parallel Multistream Training of High-Dimensional Neural Network Potentials},
author = {Andreas Singraber and Tobias Morawietz and Jörg Behler and Christoph Dellago},
doi = {10.1021/acs.jctc.8b01092},
year = {2019},
date = {2019-04-17},
journal = {Journal of Chemical Theory and Computation},
volume = {15},
number = {5},
pages = {3075--3092},
publisher = {American Chemical Society (ACS)},
abstract = {Over the past years high-dimensional neural network potentials (HDNNPs), fitted to accurately reproduce ab initio potential energy surfaces, have become a powerful tool in chemistry, physics and materials science. Here, we focus on the training of the neural networks that lies at the heart of the HDNNP method. We present an efficient approach for optimizing the weight parameters of the neural network via multistream Kalman filtering, using potential energies and forces as reference data. In this procedure, the choice of the free parameters of the Kalman filter can have a significant impact on the fit quality. Carrying out a large parameter study, we determine optimal settings and demonstrate how to optimize training results of HDNNPs. Moreover, we illustrate our HDNNP training approach by revisiting previously presented fits for water and developing a new potential for copper sulfide. This material, accessible in computer simulations so far only via first-principles methods, forms a particularly complex solid structure at low temperatures and undergoes a phase transition to a superionic state upon heating. Analyzing MD simulations carried out with the Cu_{2}S HDNNP, we confirm that the underlying ab initio reference method indeed reproduces this behavior.},
keywords = {P12, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Schlegel, Simon J; Hosseinpour, Saman; Gebhard, Maximilian; Devi, Anjana; Bonn, Mischa; Backus, Ellen H. G.
How water flips at charged titanium dioxide: an SFG-study on the water–TiO2 interface
Journal ArticleOpen AccessIn: Physical Chemistry Chemical Physics, vol. 21, no. 17, pp. 8956–8964, 2019.
Abstract | Links | BibTeX | Tags: P11, pre-TACO
@article{Schlegel2019,
title = {How water flips at charged titanium dioxide: an SFG-study on the water–TiO_{2} interface},
author = {Simon J Schlegel and Saman Hosseinpour and Maximilian Gebhard and Anjana Devi and Mischa Bonn and Ellen H. G. Backus},
doi = {10.1039/c9cp01131e},
year = {2019},
date = {2019-04-05},
urldate = {2019-04-05},
journal = {Physical Chemistry Chemical Physics},
volume = {21},
number = {17},
pages = {8956--8964},
publisher = {Royal Society of Chemistry (RSC)},
abstract = {Photocatalytic splitting of water into hydrogen and oxygen by utilizing sunlight and a photocatalyst is a promising way of generating clean energy. Here, we report a molecular-level study on heavy water (D_{2}O) interacting with TiO_{2} as a model photocatalyst. We employed the surface specific technique Sum-Frequency-Generation (SFG) spectroscopy to determine the nature of the hydrogen bonding environment and the orientation of interfacial water molecules using their OD-stretch vibrations as reporters. By examining solutions with various pD-values, we observe an intensity-minimum at around pD 5, corresponding to the balance of protonation and deprotonation of TiO_{2} (point of zero charge). The majority of water molecules’ deuterium atoms point away from the interface when the pD is below 5, and point towards the surface when the pD is higher than 5, with strong hydrogen bonds towards the surface.},
keywords = {P11, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Diebold, Ulrike; Rupprechter, Günther
Preface: Surface Science of functional oxides
Journal ArticleIn: Surface Science, vol. 681, pp. A1, 2019.
Links | BibTeX | Tags: P02, P08, pre-TACO
@article{Diebold2019,
title = {Preface: Surface Science of functional oxides},
author = {Ulrike Diebold and Günther Rupprechter},
doi = {10.1016/j.susc.2018.11.017},
year = {2019},
date = {2019-03-01},
journal = {Surface Science},
volume = {681},
pages = {A1},
publisher = {Elsevier BV},
keywords = {P02, P08, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Singraber, Andreas; Behler, Jörg; Dellago, Christoph
Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials
Journal ArticleIn: Journal of Chemical Theory and Computation, vol. 15, no. 3, pp. 1827–1840, 2019.
Abstract | Links | BibTeX | Tags: P12, pre-TACO
@article{Singraber2019a,
title = {Library-Based LAMMPS Implementation of High-Dimensional Neural Network Potentials},
author = {Andreas Singraber and Jörg Behler and Christoph Dellago},
doi = {10.1021/acs.jctc.8b00770},
year = {2019},
date = {2019-01-24},
journal = {Journal of Chemical Theory and Computation},
volume = {15},
number = {3},
pages = {1827--1840},
publisher = {American Chemical Society (ACS)},
abstract = {Neural networks and other machine learning approaches have been successfully used to accurately represent atomic interaction potentials derived from computationally demanding electronic structure calculations. Due to their low computational cost, such representations open the possibility for large scale reactive molecular dynamics simulations of processes with bonding situations that cannot be described accurately with traditional empirical force fields. Here, we present a library of functions developed for the implementation of neural network potentials. Written in C++, this library incorporates several strategies resulting in a very high efficiency of neural network potential-energy and force evaluations. Based on this library, we have developed an implementation of the neural network potential within the molecular dynamics package LAMMPS and demonstrate its performance using liquid water as a test system.},
keywords = {P12, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Reticcioli, Michele; Sokolović, Igor; Schmid, Michael; Diebold, Ulrike; Setvin, Martin; Franchini, Cesare
Interplay between Adsorbates and Polarons: CO on Rutile TiO2(110)
Journal ArticleIn: Physical Review Letters, vol. 122, no. 1, pp. 016805, 2019.
Abstract | Links | BibTeX | Tags: P02, P07, pre-TACO
@article{Reticcioli2019,
title = {Interplay between Adsorbates and Polarons: CO on Rutile TiO_{2}(110)},
author = {Michele Reticcioli and Igor Sokolović and Michael Schmid and Ulrike Diebold and Martin Setvin and Cesare Franchini},
doi = {10.1103/physrevlett.122.016805},
year = {2019},
date = {2019-01-09},
journal = {Physical Review Letters},
volume = {122},
number = {1},
pages = {016805},
publisher = {American Physical Society (APS)},
abstract = {Polaron formation plays a major role in determining the structural, electrical, and chemical properties of ionic crystals. Using a combination of first-principles calculations, scanning tunneling microscopy, and atomic force microscopy, we analyze the interaction of polarons with CO molecules adsorbed on the reduced rutile TiO_{2}(110) surface. Adsorbed CO shows attractive coupling with polarons in the surface layer, and repulsive interaction with polarons in the subsurface layer. As a result, CO adsorption depends on the reduction state of the sample. For slightly reduced surfaces, many adsorption configurations with comparable adsorption energies exist and polarons reside in the subsurface layer. At strongly reduced surfaces, two adsorption configurations dominate: either inside an oxygen vacancy, or at surface Ti_{5c} sites, coupled with a surface polaron. Similar conclusions are predicted for TiO_{2}(110) surfaces containing near-surface Ti interstitials. These results show that polarons are of primary importance for understanding the performance of polar semiconductors and transition metal oxides in catalysis and energy-related applications.},
keywords = {P02, P07, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Cheng, Bingqing; Engel, Edgar A; Behler, Jörg; Dellago, Christoph; Ceriotti, Michele
Ab initio thermodynamics of liquid and solid water
Journal ArticleOpen AccessIn: Proceedings of the National Academy of Sciences, vol. 116, no. 4, pp. 1110–1115, 2019.
Abstract | Links | BibTeX | Tags: P12, pre-TACO
@article{Cheng2019,
title = {Ab initio thermodynamics of liquid and solid water},
author = {Bingqing Cheng and Edgar A Engel and Jörg Behler and Christoph Dellago and Michele Ceriotti},
doi = {10.1073/pnas.1815117116},
year = {2019},
date = {2019-01-04},
urldate = {2019-01-04},
journal = {Proceedings of the National Academy of Sciences},
volume = {116},
number = {4},
pages = {1110--1115},
publisher = {Proceedings of the National Academy of Sciences},
abstract = {A central goal of computational physics and chemistry is to predict material properties by using first-principles methods based on the fundamental laws of quantum mechanics. However, the high computational costs of these methods typically prevent rigorous predictions of macroscopic quantities at finite temperatures, such as heat capacity, density, and chemical potential. Here, we enable such predictions by marrying advanced free-energy methods with data-driven machine-learning interatomic potentials. We show that, for the ubiquitous and technologically essential system of water, a first-principles thermodynamic description not only leads to excellent agreement with experiments, but also reveals the crucial role of nuclear quantum fluctuations in modulating the thermodynamic stabilities of different phases of water.},
keywords = {P12, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
2018
Lukashuk, Liliana; Yigit, Nevzat; Rameshan, Raffael; Kolar, Elisabeth; Teschner, Detre; Hävecker, Michael; Knop-Gericke, Axel; Schlögl, Robert; Föttinger, Karin; Rupprechter, Günther
Operando Insights into CO Oxidation on Cobalt Oxide Catalysts by NAP-XPS, FTIR, and XRD
Journal ArticleOpen AccessIn: ACS Catalysis, vol. 8, no. 9, pp. 8630–8641, 2018.
Abstract | Links | BibTeX | Tags: P08, P10, pre-TACO
@article{Lukashuk2018,
title = {Operando Insights into CO Oxidation on Cobalt Oxide Catalysts by NAP-XPS, FTIR, and XRD},
author = {Liliana Lukashuk and Nevzat Yigit and Raffael Rameshan and Elisabeth Kolar and Detre Teschner and Michael Hävecker and Axel Knop-Gericke and Robert Schlögl and Karin Föttinger and Günther Rupprechter},
doi = {10.1021/acscatal.8b01237},
year = {2018},
date = {2018-08-07},
urldate = {2018-08-07},
journal = {ACS Catalysis},
volume = {8},
number = {9},
pages = {8630--8641},
publisher = {American Chemical Society (ACS)},
abstract = {Cobalt oxide Co_{3}O_{4} has recently emerged as promising, noble metal-free catalyst for oxidation reactions but a better understanding of the active catalyst under working conditions is required for further development and potential commercialization. An operando approach has been applied, combining near ambient (atmospheric) pressure X-ray photoelectron spectroscopy (NAP-XPS), Fourier transform infrared spectroscopy (FTIR), or X-ray diffraction (XRD) with simultaneous catalytic tests of CO oxidation on Co_{3}O_{4}, enabling one to monitor surface and bulk states under various reaction conditions (steady-state and dynamic conditions switching between CO and O_{2}). On the basis of the surface-specific chemical information a complex network of different reaction pathways unfolded: Mars-van-Krevelen (MvK), CO dissociation followed by carbon oxidation, and formation of carbonates. A possible Langmuir–Hinshelwood (LH) pathway cannot be excluded because of the good activity when no oxygen vacancies were detected. The combined NAP-XPS/FTIR results are in line with a MvK mechanism above 100 °C, involving the Co^{3+}/Co^{2+} redox couple and oxygen vacancy formation. Under steady state, the Co_{3}O_{4} surface appeared oxidized and the amount of reduced Co^{2+} species at/near the surface remained low up to 200 °C. Only in pure CO, about 15% of surface reduction were detected, suggesting that the active sites are a minority species. The operando spectroscopic studies also revealed additional reaction pathways: CO dissociation followed by carbon reoxidation and carbonate formation and its decomposition. However, due to their thermal stability in various atmospheres, the carbonates are rather spectators and also CO dissociation seems a minor route. This study thus highlights the benefits of combining operando surface sensitive techniques to gain insight into catalytically active surfaces.},
keywords = {P08, P10, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
2017
Reticcioli, Michele; Setvin, Martin; Hao, Xianfeng; Flauger, Peter; Kresse, Georg; Schmid, Michael; Diebold, Ulrike; Franchini, Cesare
Polaron-Driven Surface Reconstructions
Journal ArticleOpen AccessIn: Physical Review X, vol. 7, no. 3, pp. 031053, 2017.
Abstract | Links | BibTeX | Tags: P02, P03, P07, pre-TACO
@article{Reticcioli2017,
title = {Polaron-Driven Surface Reconstructions},
author = {Michele Reticcioli and Martin Setvin and Xianfeng Hao and Peter Flauger and Georg Kresse and Michael Schmid and Ulrike Diebold and Cesare Franchini},
doi = {10.1103/physrevx.7.031053},
year = {2017},
date = {2017-09-25},
urldate = {2017-09-25},
journal = {Physical Review X},
volume = {7},
number = {3},
pages = {031053},
publisher = {American Physical Society (APS)},
abstract = {Geometric and electronic surface reconstructions determine the physical and chemical properties of surfaces and, consequently, their functionality in applications. The reconstruction of a surface minimizes its surface free energy in otherwise thermodynamically unstable situations, typically caused by dangling bonds, lattice stress, or a divergent surface potential, and it is achieved by a cooperative modification of the atomic and electronic structure. Here, we combined first-principles calculations and surface techniques (scanning tunneling microscopy, non-contact atomic force microscopy, scanning tunneling spectroscopy) to report that the repulsion between negatively charged polaronic quasiparticles, formed by the interaction between excess electrons and the lattice phonon field, plays a key role in surface reconstructions. As a paradigmatic example, we explain the (1×1) to (1×2) transition in rutile TiO_{2}(110).},
keywords = {P02, P03, P07, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
Legrain, Fleur; Carrete, Jesús; van Roekeghem, Ambroise; Curtarolo, Stefano; Mingo, Natalio
How Chemical Composition Alone Can Predict Vibrational Free Energies and Entropies of Solids
Journal ArticleIn: Chemistry of Materials, vol. 29, no. 15, pp. 6220–6227, 2017.
Abstract | Links | BibTeX | Tags: P09, pre-TACO
@article{Legrain2017,
title = {How Chemical Composition Alone Can Predict Vibrational Free Energies and Entropies of Solids},
author = {Fleur Legrain and Jesús Carrete and Ambroise van Roekeghem and Stefano Curtarolo and Natalio Mingo},
doi = {10.1021/acs.chemmater.7b00789},
year = {2017},
date = {2017-06-22},
journal = {Chemistry of Materials},
volume = {29},
number = {15},
pages = {6220--6227},
publisher = {American Chemical Society (ACS)},
abstract = {Computing vibrational free energies (F_{vib}) and entropies (S_{vib}) has posed a long-standing challenge to the high-throughput ab initio investigation of finite temperature properties of solids. Here, we use machine-learning techniques to efficiently predict F_{vib} and S_{vib} of crystalline compounds in the Inorganic Crystal Structure Database. Using descriptors based simply on the chemical formula and using a training set of only 300 compounds, mean absolute errors of less than 0.04 meV/K/atom (15 meV/atom) are achieved for S_{vib} (F_{vib}), whose values are distributed within a range of 0.9 meV/K/atom (300 meV/atom.) In addition, for training sets containing fewer than 2000 compounds, the chemical formula alone is shown to perform as well as, if not better than, four other more complex descriptors previously used in the literature. The accuracy and simplicity of the approach means that it can be advantageously used for fast screening of chemical reactions at finite temperatures.},
keywords = {P09, pre-TACO},
pubstate = {published},
tppubtype = {article}
}
2016
Lukashuk, Liliana; Föttinger, Karin; Kolar, Elisabeth; Rameshan, Christoph; Teschner, Detre; Hävecker, Michael; Knop-Gericke, Axel; Yigit, Nevzat; Li, Hao; McDermott, Eamon; Stöger-Pollach, Michael; Rupprechter, Günther
Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts
Journal ArticleOpen AccessIn: Journal of Catalysis, vol. 344, pp. 1–15, 2016.
Abstract | Links | BibTeX | Tags: P08, P10, pre-TACO
@article{Lukashuk2016,
title = {Operando XAS and NAP-XPS studies of preferential CO oxidation on Co_{3}O_{4} and CeO_{2}-Co_{3}O_{4 }catalysts},
author = {Liliana Lukashuk and Karin Föttinger and Elisabeth Kolar and Christoph Rameshan and Detre Teschner and Michael Hävecker and Axel Knop-Gericke and Nevzat Yigit and Hao Li and Eamon McDermott and Michael Stöger-Pollach and Günther Rupprechter},
doi = {10.1016/j.jcat.2016.09.002},
year = {2016},
date = {2016-12-01},
urldate = {2016-12-01},
journal = {Journal of Catalysis},
volume = {344},
pages = {1--15},
publisher = {Elsevier BV},
abstract = {Co_{3}O_{4} is a promising catalyst for removing CO from H_{2} streams via the preferential CO oxidation (PROX). A Mars-van-Krevelen redox mechanism is often suggested but a detailed knowledge especially of the oxidation state of the catalytically active surface under reaction conditions is typically missing. We have thus utilized operando X-ray absorption spectroscopy to examine structure and oxidation state during PROX, and near atmospheric pressure-XPS at low photoelectron kinetic energies and thus high surface sensitivity to monitor surface composition changes. The rather easy surface reduction in pure CO (starting already at ∼100 °C) and the easy reoxidation by O_{2} suggest that molecularly adsorbed CO reacts with lattice oxygen, which is replenished by gas phase O_{2}. Nevertheless, the steady state concentration of oxygen vacancies under reaction conditions is too low even for XPS detection so that both the bulk and surface of Co_{3}O_{4} appear fully oxidized during PROX. Furthermore, the effect of adding CeO_{2} (a less active material) to Co_{3}O_{4} was studied. Promotion of Co_{3}O_{4} with 10 wt% CeO_{2} increases the reduction temperatures in CO and H_{2} and enhances the PROX activity. Since CeO_{2} is a less active material, this can only be explained by a higher activity of the Co-O-Ce interface.},
keywords = {P08, P10, pre-TACO},
pubstate = {published},
tppubtype = {article}
}